

A novel clustering method via nucleotide-based Fourier power spectrum analysis
- PMID: 21443881
- PMCID: PMC7094093
- DOI: 10.1016/j.jtbi.2011.03.029
A novel clustering method via nucleotide-based Fourier power spectrum analysis
Abstract
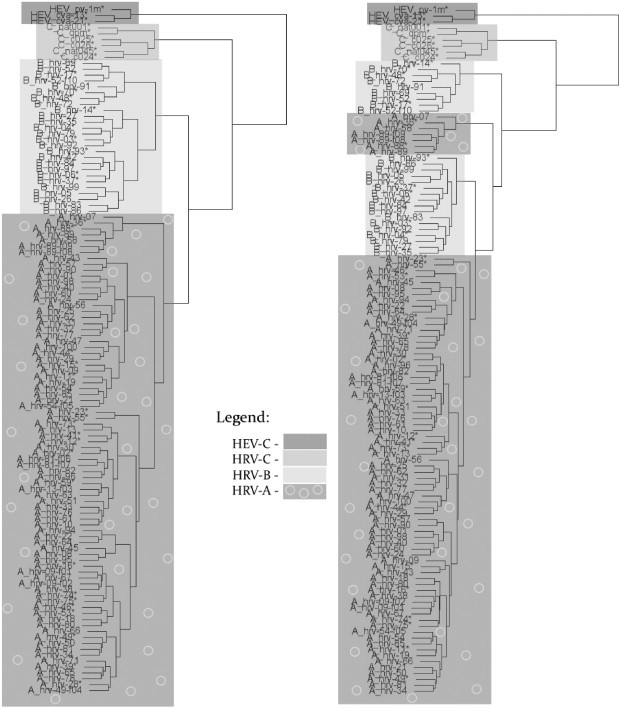
A novel clustering method is proposed to classify genes or genomes. This method uses a natural representation of genomic data by binary indicator sequences of each nucleotide (adenine (A), cytosine (C), guanine (G), and thymine (T)). Afterwards, the discrete Fourier transform is applied to these indicator sequences to calculate spectra of the nucleotides. Mathematical moments are calculated for each of these spectra to create a multidimensional vector in a Euclidean space for each gene or genome sequence. Thus, each gene or genome sequence is realized as a geometric point in the Euclidean space. Finally, pairwise Euclidean distances between these points (i.e. genome sequences) are calculated to cluster the gene or genome sequences. This method is applied to three sets of data. The first is 34 strains of coronavirus genomic data, the second is 118 of the known strains of Human rhinovirus (HRV), and the third is 30 bacteria genomes. The distance matrices are computed based on the three sets, showing the distances from each point to the others. We used the complete linkage clustering algorithm to build phylogenetic trees to indicate how the distances among these sequence correspond to the evolutionary relationship among these sequences. This genome representation provides a powerful and efficient method to classify genomes and is much faster than the widely acknowledged multiple sequence alignment method.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures


References
-
- Brown N.P., Larkin M.A., Blackshields G. Clustal w and clustal x version 2.0. Bioinformatics. 2007;23(21):2947–2948. - PubMed
-
- Dawyndt P., De Meyer H., De Baets B. The complete linkage clustering algorithm revisited. Soft Computing. 2005;9(May):385–392.
-
- Katoh K., Asimenos G., Toh H. Multiple alignment of dna sequences with MAFFT. Methods Molecular Biology. 2009;537:39–64. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
