

Corticotropin releasing factor-induced CREB activation in striatal neurons occurs via a novel Gβγ signaling pathway
- PMID: 21448293
- PMCID: PMC3063246
- DOI: 10.1371/journal.pone.0018114
Corticotropin releasing factor-induced CREB activation in striatal neurons occurs via a novel Gβγ signaling pathway
Abstract
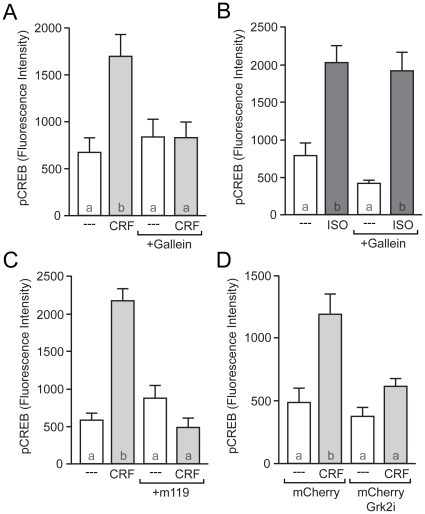
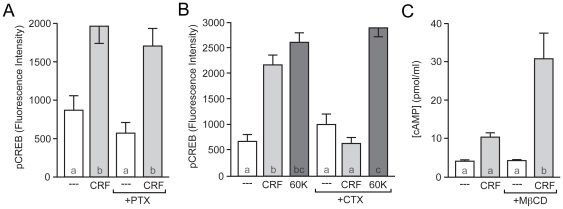
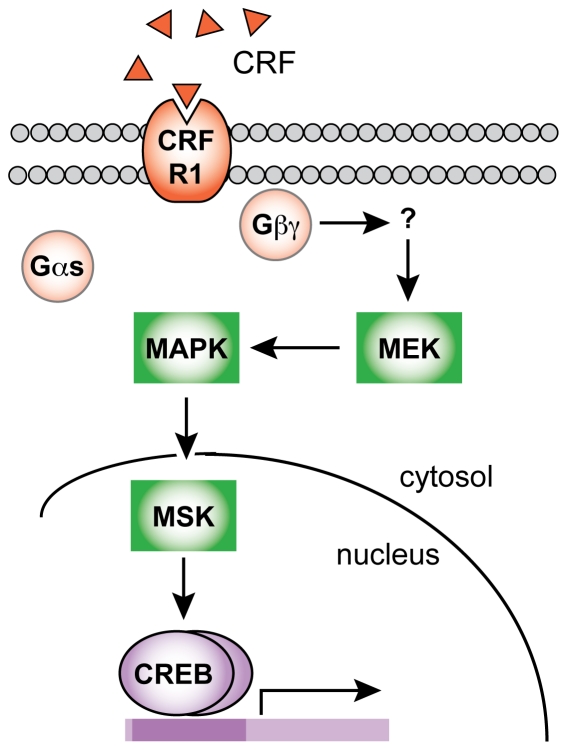
The peptide corticotropin-releasing factor (CRF) was initially identified as a critical component of the stress response. CRF exerts its cellular effects by binding to one of two cognate G-protein coupled receptors (GPCRs), CRF receptor 1 (CRFR1) or 2 (CRFR2). While these GPCRs were originally characterized as being coupled to Gα(s), leading to downstream activation of adenylyl cyclase (AC) and subsequent increases in cAMP, it has since become clear that CRFRs couple to and activate numerous other downstream signaling cascades. In addition, CRF signaling influences the activity of many diverse brain regions, affecting a variety of behaviors. One of these regions is the striatum, including the nucleus accumbens (NAc). CRF exerts profound effects on striatal-dependent behaviors such as drug addiction, pair-bonding, and natural reward. Recent data indicate that at least some of these behaviors regulated by CRF are mediated through CRF activation of the transcription factor CREB. Thus, we aimed to elucidate the signaling pathway by which CRF activates CREB in striatal neurons. Here we describe a novel neuronal signaling pathway whereby CRF leads to a rapid Gβγ- and MEK-dependent increase in CREB phosphorylation. These data are the first descriptions of CRF leading to activation of a Gβγ-dependent signaling pathway in neurons, as well as the first description of Gβγ activation leading to downstream CREB phosphorylation in any cellular system. Additionally, these data provide additional insight into the mechanisms by which CRF can regulate neuronal function.
Conflict of interest statement
Figures
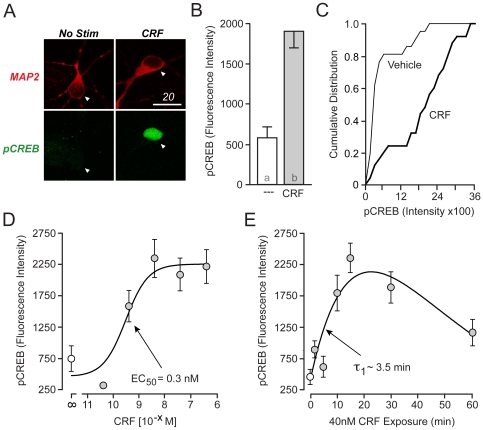
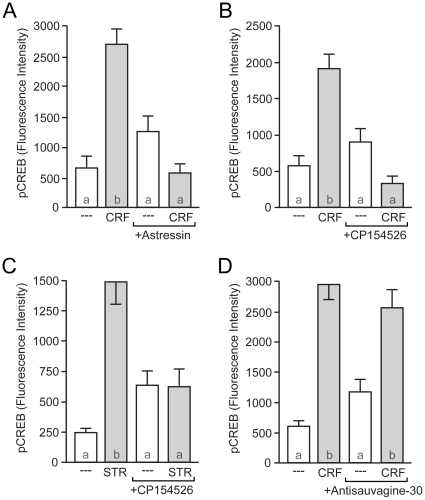
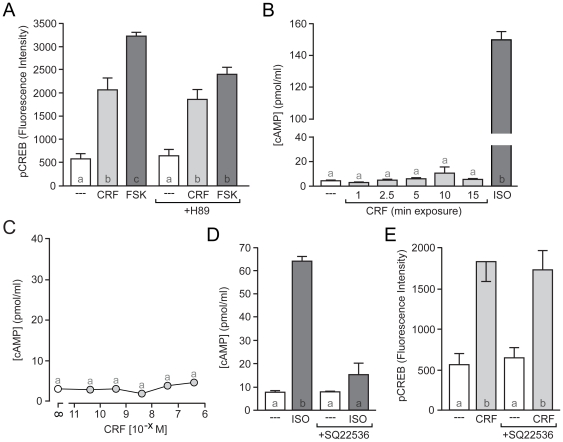
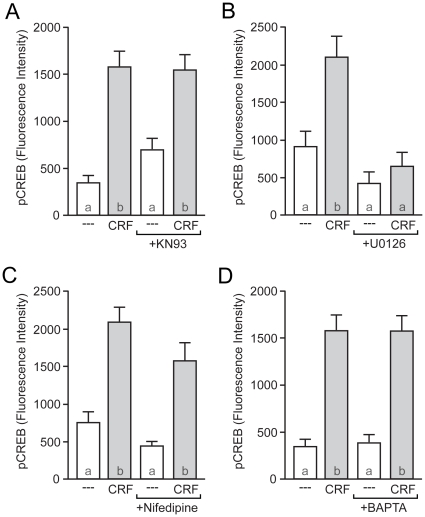
Similar articles
-
Corticotropin-releasing factor and urocortin I activate CREB through functionally selective Gβγ signaling in hippocampal pyramidal neurons.Eur J Neurosci. 2011 Sep;34(5):671-81. doi: 10.1111/j.1460-9568.2011.07812.x. Epub 2011 Aug 8. Eur J Neurosci. 2011. PMID: 21819464 Free PMC article.
-
Corticotropin-releasing factor type 1 and type 2alpha receptors regulate phosphorylation of calcium/cyclic adenosine 3',5'-monophosphate response element-binding protein and activation of p42/p44 mitogen-activated protein kinase.Endocrinology. 1999 Apr;140(4):1525-36. doi: 10.1210/endo.140.4.6656. Endocrinology. 1999. PMID: 10098484
-
Differential regulation of CREB and ERK phosphorylation through corticotropin-releasing factor receptors type 1 and 2 in AtT-20 and A7r5 cells.Mol Cell Endocrinol. 2007 Jan 15;263(1-2):90-102. doi: 10.1016/j.mce.2006.08.011. Epub 2006 Oct 5. Mol Cell Endocrinol. 2007. PMID: 17027144
-
Reflections on: "A general role for adaptations in G-Proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function".Brain Res. 2016 Aug 15;1645:71-4. doi: 10.1016/j.brainres.2015.12.039. Epub 2015 Dec 29. Brain Res. 2016. PMID: 26740398 Free PMC article. Review.
-
Alteration of second messengers during acute cerebral ischemia - adenylate cyclase, cyclic AMP-dependent protein kinase, and cyclic AMP response element binding protein.Prog Neurobiol. 2001 Oct;65(2):173-207. doi: 10.1016/s0301-0082(01)00002-8. Prog Neurobiol. 2001. PMID: 11403878 Review.
Cited by
-
Nerve growth factor induces neurite outgrowth of PC12 cells by promoting Gβγ-microtubule interaction.BMC Neurosci. 2014 Dec 31;15:132. doi: 10.1186/s12868-014-0132-4. BMC Neurosci. 2014. PMID: 25552352 Free PMC article.
-
Involvement of Akt/CREB signaling pathways in the protective effect of EPA against interleukin-1β-induced cytotoxicity and BDNF down-regulation in cultured rat hippocampal neurons.BMC Neurosci. 2018 Sep 6;19(1):52. doi: 10.1186/s12868-018-0455-7. BMC Neurosci. 2018. PMID: 30189852 Free PMC article.
-
Endocrinology and the brain: corticotropin-releasing hormone signaling.Endocr Connect. 2017 Aug;6(6):R99-R120. doi: 10.1530/EC-17-0111. Epub 2017 Jul 14. Endocr Connect. 2017. PMID: 28710078 Free PMC article. Review.
-
Genome-Wide Identification of Basic Helix-Loop-Helix and NF-1 Motifs Underlying GR Binding Sites in Male Rat Hippocampus.Endocrinology. 2017 May 1;158(5):1486-1501. doi: 10.1210/en.2016-1929. Endocrinology. 2017. PMID: 28200020 Free PMC article.
-
Early Life Stress Associated With Increased Striatal N-Acetyl-Aspartate: Cerebrospinal Fluid Corticotropin-Releasing Factor Concentrations, Hippocampal Volume, Body Mass and Behavioral Correlates.Chronic Stress (Thousand Oaks). 2018 Jan-Dec;2:2470547018768450. doi: 10.1177/2470547018768450. Epub 2018 Apr 18. Chronic Stress (Thousand Oaks). 2018. PMID: 29963652 Free PMC article.
References
-
- Sarnyai Z. Neurobiology of stress and cocaine addiction. Studies on corticotropin-releasing factor in rats, monkeys, and humans. Ann N Y Acad Sci. 1998;851:371–387. - PubMed
-
- Brown SA, Vik PW, Patterson TL, Grant I, Schuckit MA. Stress, vulnerability and adult alcohol relapse. J Stud Alcohol. 1995;56:538–545. - PubMed
-
- Shaham Y, Erb S, Stewart J. Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res Brain Res Rev. 2000;33:13–33. - PubMed
-
- Goeders NE, Guerin GF. Non-contingent electric footshock facilitates the acquisition of intravenous cocaine self-administration in rats. Psychopharmacology (Berl) 1994;114:63–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
