

Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10
- PMID: 21448667
- PMCID: PMC6959532
- DOI: 10.1007/s12013-011-9179-3
Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10
Abstract
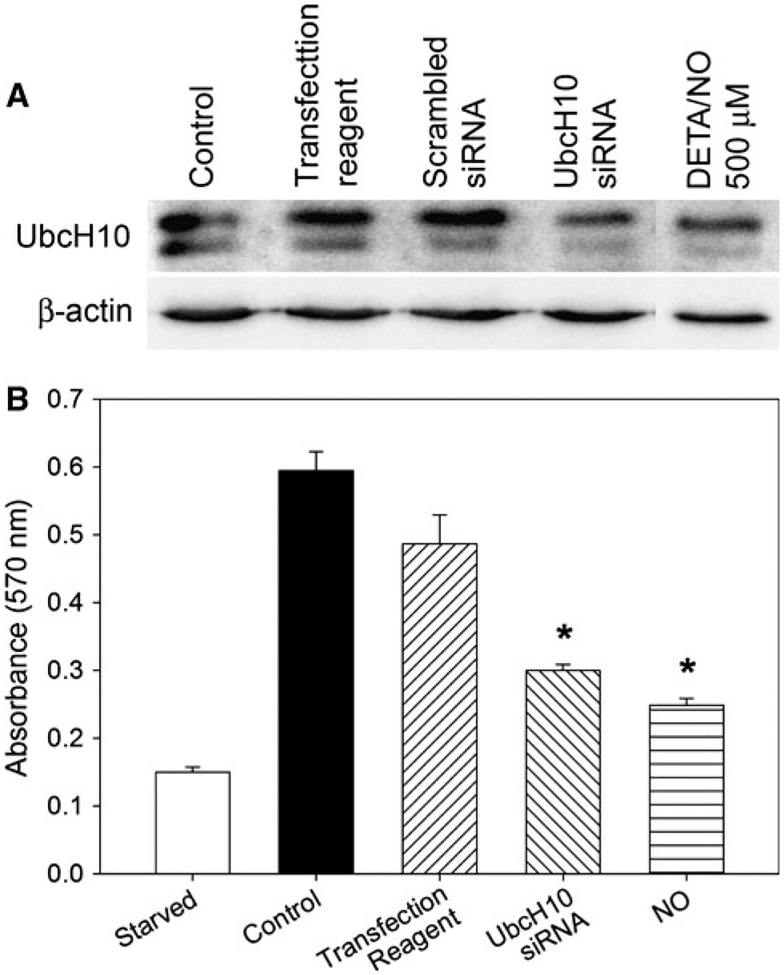
Nitric oxide (NO) limits formation of neointimal hyperplasia in animal models of arterial injury in large part by inhibiting vascular smooth muscle cell (VSMC) proliferation through cell cycle arrest. The ubiquitin-conjugating enzyme UbcH10 is responsible for ubiquitinating cell cycle proteins for proper exit from mitosis. We hypothesize that NO prevents VSMC proliferation, and hence neointimal hyperplasia, by decreasing levels of UbcH10. Western blotting and immunofluorescent staining showed that NO reduced UbcH10 levels in a concentration-dependent manner in VSMC harvested from the abdominal aortas of Sprague-Dawley rats. Treatment with NO or siRNA to UbcH10 decreased both UbcH10 levels and VSMC proliferation (P<0.001), while increasing UbcH10 levels by plasmid transfection or angiotensin II stimulation increased VSMC proliferation to 150% (P=0.008) and 212% (P=0.002) of control, respectively. Immunofluorescent staining of balloon-injured rat carotid arteries showed a ~4-fold increase in UbcH10 levels, which was profoundly decreased following treatment with NO. Western blotting of carotid artery lysates showed no UbcH10 in uninjured vessels, a substantial increase in the injury alone group, and a significant decrease in the injury+NO group (~3-fold reduction versus injury alone). Importantly, in vitro and in vivo, a marked increase in polyubiquitinated UbcH10 was observed in the NO-treated VSMC and carotid arteries, respectively, indicating that NO may be decreasing unmodified UbcH10 levels by increasing its ubiquitination. Central to our hypothesis, we report that NO decreases UbcH10 levels in VSMC in vitro and following arterial injury in vivo in association with increasing polyubiquitinated-UbcH10 levels. These changes in UbcH10 levels correlate with VSMC proliferation and neointimal hyperplasia, making UbcH10 a promising therapeutic target for inhibiting this proliferative disease.
Figures






Similar articles
-
Nitric oxide and nanotechnology: a novel approach to inhibit neointimal hyperplasia.J Vasc Surg. 2008 Jan;47(1):173-82. doi: 10.1016/j.jvs.2007.09.005. J Vasc Surg. 2008. PMID: 18178471 Free PMC article.
-
Isopropylamine NONOate (IPA/NO) moderates neointimal hyperplasia following vascular injury.J Vasc Surg. 2010 May;51(5):1248-59. doi: 10.1016/j.jvs.2009.12.028. Epub 2010 Mar 11. J Vasc Surg. 2010. PMID: 20223627 Free PMC article.
-
Effect of nitric oxide on neointimal hyperplasia based on sex and hormone status.Free Radic Biol Med. 2011 May 1;50(9):1065-74. doi: 10.1016/j.freeradbiomed.2011.01.016. Epub 2011 Jan 21. Free Radic Biol Med. 2011. PMID: 21256959 Free PMC article.
-
Nitric oxide affects UbcH10 levels differently in type 1 and type 2 diabetic rats.J Surg Res. 2015 Jun 1;196(1):180-9. doi: 10.1016/j.jss.2015.02.012. Epub 2015 Feb 13. J Surg Res. 2015. PMID: 25801975 Free PMC article.
-
Heightened efficacy of nitric oxide-based therapies in type II diabetes mellitus and metabolic syndrome.Am J Physiol Heart Circ Physiol. 2008 Dec;295(6):H2388-98. doi: 10.1152/ajpheart.00185.2008. Epub 2008 Oct 17. Am J Physiol Heart Circ Physiol. 2008. PMID: 18931034 Free PMC article.
Cited by
-
Endothelial dysfunction: molecular mechanisms and clinical implications.MedComm (2020). 2024 Jul 22;5(8):e651. doi: 10.1002/mco2.651. eCollection 2024 Aug. MedComm (2020). 2024. PMID: 39040847 Free PMC article. Review.
-
Proteomics Studies Suggest That Nitric Oxide Donor Furoxans Inhibit In Vitro Vascular Smooth Muscle Cell Proliferation by Nitric Oxide-Independent Mechanisms.Molecules. 2023 Jul 28;28(15):5724. doi: 10.3390/molecules28155724. Molecules. 2023. PMID: 37570694 Free PMC article.
-
Why is coronary collateral growth impaired in type II diabetes and the metabolic syndrome?Vascul Pharmacol. 2012 Nov-Dec;57(5-6):179-86. doi: 10.1016/j.vph.2012.02.001. Epub 2012 Feb 9. Vascul Pharmacol. 2012. PMID: 22342811 Free PMC article. Review.
-
Novel Therapeutics for Type 2 Diabetes Mellitus-A Look at the Past Decade and a Glimpse into the Future.Biomedicines. 2024 Jun 21;12(7):1386. doi: 10.3390/biomedicines12071386. Biomedicines. 2024. PMID: 39061960 Free PMC article. Review.
-
Nitric oxide increases lysine 48-linked ubiquitination following arterial injury.J Surg Res. 2011 Sep;170(1):e169-77. doi: 10.1016/j.jss.2011.05.032. Epub 2011 Jun 15. J Surg Res. 2011. PMID: 21737094 Free PMC article.
References
-
- Rape M, & Kirschner MW (2004). Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature, 432(7017), 588–595. - PubMed
-
- Rape M, Reddy SK, & Kirschner MW (2006). The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell, 124(1), 89–103. - PubMed
-
- Hershko A, & Ciechanover A (1998). The ubiquitin system. Annual Review of Biochemistry, 67, 425–479. - PubMed
-
- Crews CM (2003). Feeding the machine: Mechanisms of proteasome-catalyzed degradation of ubiquitinated proteins. Current Opinion in Chemical Biology, 7(5), 534–539. - PubMed
-
- De GA, Ganier O, & Cohen-Fix O (2006). Before and after the spindle assembly checkpoint—an APC/C point of view. Cell Cycle, 5(18), 2168–2171. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
