

Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype
- PMID: 21454527
- PMCID: PMC3089578
- DOI: 10.1074/jbc.M110.192682
Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype
Abstract
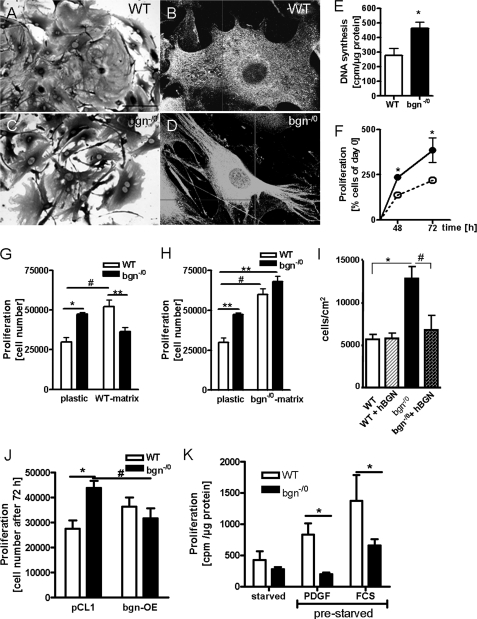
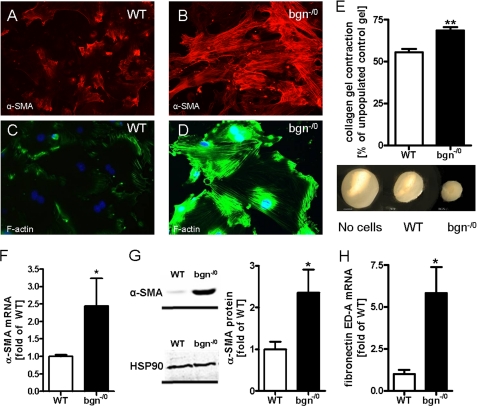
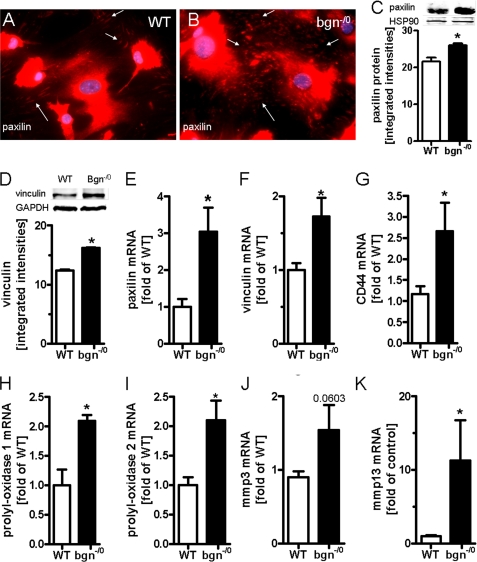
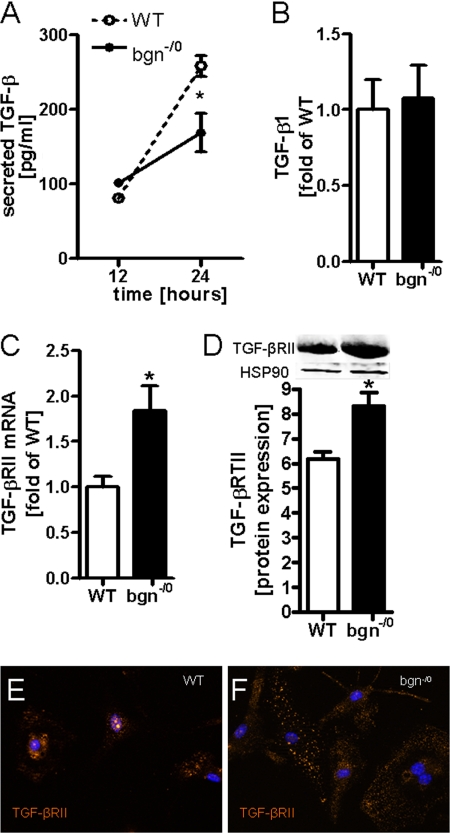
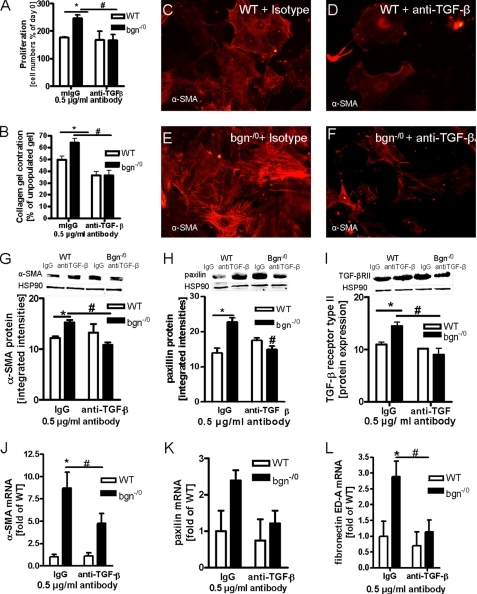
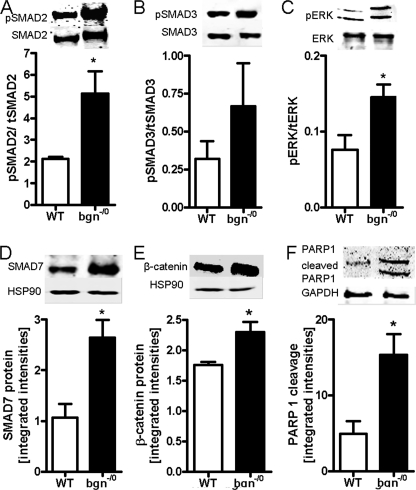
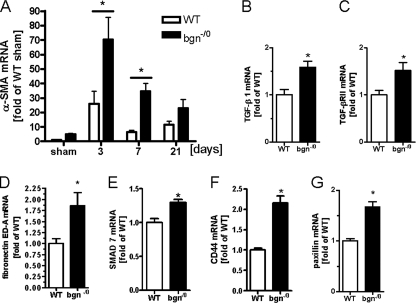
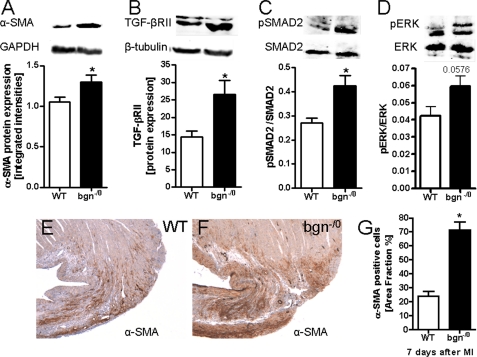
Myocardial infarction (MI) is followed by extracellular matrix (ECM) remodeling, which is on the one hand required for the healing response and the formation of stable scar tissue. However, on the other hand, ECM remodeling can lead to fibrosis and decreased ventricular compliance. The small leucine-rich proteoglycan (SLRP), biglycan (bgn), has been shown to be critically involved in these processes. During post-infarct remodeling cardiac fibroblasts differentiate into myofibroblasts which are the main cell type mediating ECM remodeling. The aim of the present study was to characterize the role of bgn in modulating the phenotype of cardiac fibroblasts. Cardiac fibroblasts were isolated from hearts of wild-type (WT) versus bgn(-/0) mice. Phenotypic characterization of the bgn(-/0) fibroblasts revealed increased proliferation. Importantly, this phenotype of bgn(-/0) fibroblasts was abolished to the WT level by reconstitution of biglycan in the ECM. TGF-β receptor II expression and phosphorylation of SMAD2 were increased. Furthermore, indicative of a myofibroblast phenotype bgn(-/0) fibroblasts were characterized by increased α-smooth muscle actin (α-SMA) incorporated into stress fibers, increased formation of focal adhesions, and increased contraction of collagen gels. Administration of neutralizing antibodies to TGF-β reversed the pro-proliferative, myofibroblastic phenotype. In vivo post-MI α-SMA, TGF-β receptor II expression, and SMAD2 phosphorylation were markedly increased in bgn(-/0) mice. Collectively, the data suggest that bgn deficiency promotes myofibroblast differentiation and proliferation in vitro and in vivo likely due to increased responses to TGF-β and SMAD2 signaling.
Figures
References
-
- Berschneider H. M. (1992) Ann. N. Y. Acad. Sci. 664, 140–147 - PubMed
-
- Hinterleitner T. A., Saada J. I., Berschneider H. M., Powell D. W., Valentich J. D. (1996) Am. J. Physiol. 271, C1262–C1268 - PubMed
-
- Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., Brown R. A. (2002) Nat. Rev. Mol. Cell Biol. 3, 349–363 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
