

Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation
- PMID: 21454558
- PMCID: PMC3091214
- DOI: 10.1074/jbc.M111.223602
Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation
Abstract
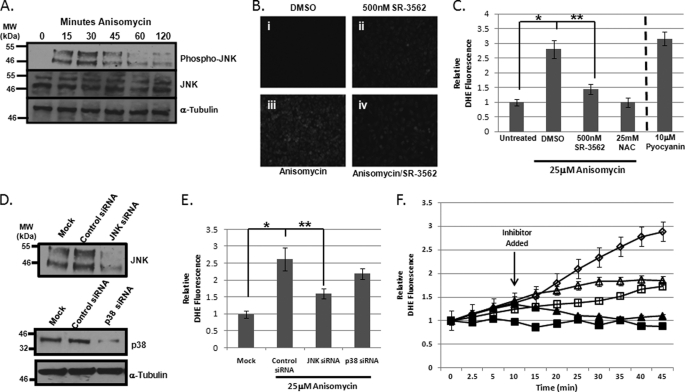
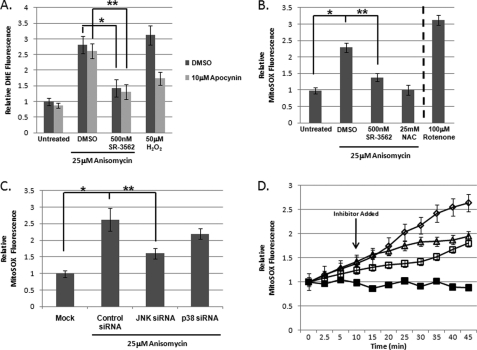
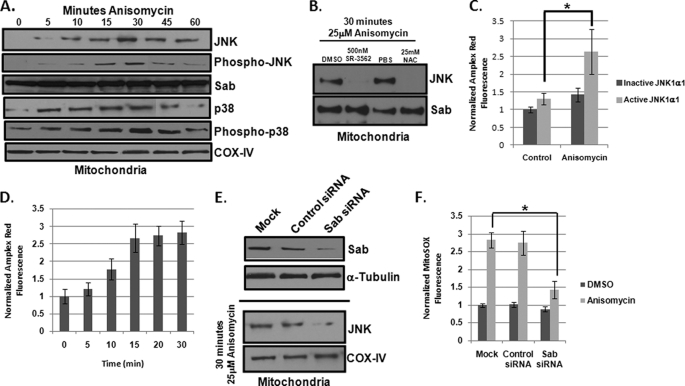
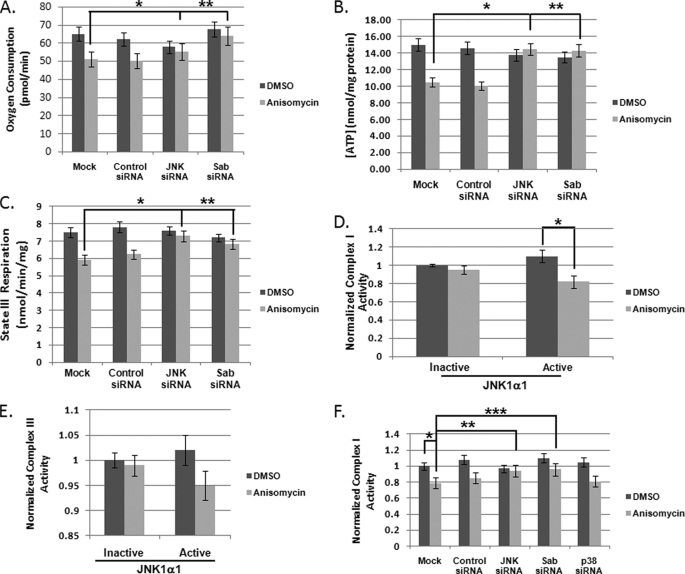
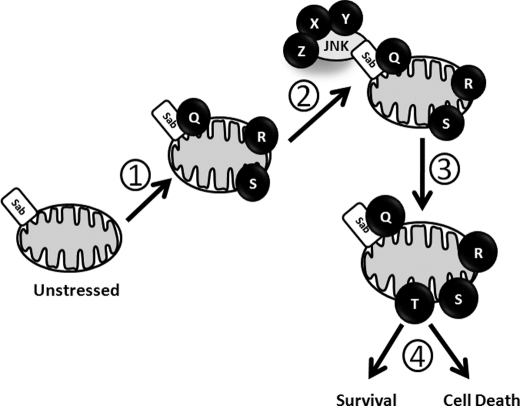
The JNK signaling cascade is critical for cellular responses to a variety of environmental and cellular stimuli. Although gene expression aspects of JNK signal transduction are well studied, there are minimal data on the physiological impact of JNK signaling. To bridge this gap, we investigated how JNK impacted physiology in HeLa cells. We observed that inhibition of JNK activity and JNK silencing with siRNA reduced the level of reactive oxygen species (ROS) generated during anisomycin-induced stress in HeLa cells. Silencing p38 had no significant impact on ROS generation under anisomycin stress. Moreover, JNK signaling mediated amplification of ROS production during stress. Mitochondrial superoxide production was shown to be the source of JNK-induced ROS amplification, as an NADPH oxidase inhibitor demonstrated little impact on JNK-mediated ROS generation. Using mitochondrial isolation from JNK null fibroblasts and targeting the mitochondrial scaffold of JNK, Sab, we demonstrated that mitochondrial JNK signaling was responsible for mitochondrial superoxide amplification. These results suggest that cellular stress altered mitochondria, causing JNK to translocate to the mitochondria and amplify up to 80% of the ROS generated largely by Complex I. This work demonstrates that a sequence of events exist for JNK mitochondrial signaling whereby ROS activates JNK, thereby affecting mitochondrial physiology, which can have effects on cell survival and death.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
