

Genomic deletions in OPA1 in Danish patients with autosomal dominant optic atrophy
- PMID: 21457585
- PMCID: PMC3079616
- DOI: 10.1186/1471-2350-12-49
Genomic deletions in OPA1 in Danish patients with autosomal dominant optic atrophy
Abstract
Background: Autosomal dominant optic atrophy (ADOA, Kjer disease, MIM #165500) is the most common form of hereditary optic neuropathy. Mutations in OPA1 located at chromosome 3q28 are the predominant cause for ADOA explaining between 32 and 89% of cases. Although deletions of OPA1 were recently reported in ADOA, the frequency of OPA1 genomic rearrangements in Denmark, where ADOA has a high prevalence, is unknown. The aim of the study was to identify copy number variations in OPA1 in Danish ADOA patients.
Methods: Forty unrelated ADOA patients, selected from a group of 100 ADOA patients as being negative for OPA1 point mutations, were tested for genomic rearrangements in OPA1 by multiplex ligation probe amplification (MLPA). When only one probe was abnormal results were confirmed by additional manually added probes. Segregation analysis was performed in families with detected mutations when possible.
Results: Ten families had OPA1 deletions, including two with deletions of the entire coding region and eight with intragenic deletions. Segregation analysis was possible in five families, and showed that the deletions segregated with the disease.
Conclusion: Deletions in the OPA1 gene were found in 10 patients presenting with phenotypic autosomal dominant optic neuropathy. Genetic testing for deletions in OPA1 should be offered for patients with clinically diagnosed ADOA and no OPA1 mutations detected by DNA sequencing analysis.
Figures
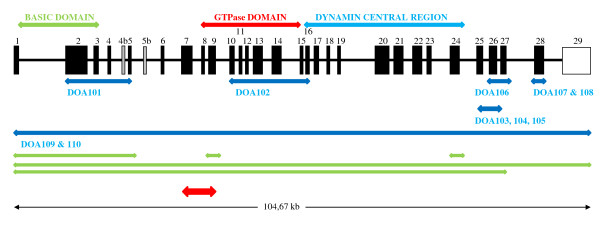
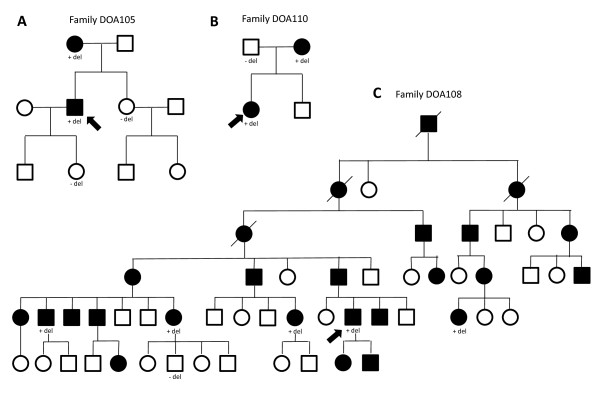
References
-
- Kerrison JB. Hereditary optic neuropathies. Ophthalmol Clin North Am. 2001;14:99–107. - PubMed
-
- Kjer P. Infantile optic atrophy with dominant mode of inheritance: a clinical and genetic study of 19 Danish families. Acta Ophthalmol Suppl. 1959;164:1–147. - PubMed
-
- Thiselton DL, Alexander C, Morris A, Brooks S, Rosenberg T, Eiberg H, Kjer B, Kjer P, Bhattacharya SS, Votruba M. A frameshift mutation in exon 28 of the OPA1 gene explains the high prevalence of dominant optic atrophy in the Danish population: evidence for a founder effect. Hum Genet. 2001;109:498–502. doi: 10.1007/s004390100600. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
