

A genome-wide comparison of the functional properties of rare and common genetic variants in humans
- PMID: 21457907
- PMCID: PMC3071924
- DOI: 10.1016/j.ajhg.2011.03.008
A genome-wide comparison of the functional properties of rare and common genetic variants in humans
Abstract
One of the longest running debates in evolutionary biology concerns the kind of genetic variation that is primarily responsible for phenotypic variation in species. Here, we address this question for humans specifically from the perspective of population allele frequency of variants across the complete genome, including both coding and noncoding regions. We establish simple criteria to assess the likelihood that variants are functional based on their genomic locations and then use whole-genome sequence data from 29 subjects of European origin to assess the relationship between the functional properties of variants and their population allele frequencies. We find that for all criteria used to assess the likelihood that a variant is functional, the rarer variants are significantly more likely to be functional than the more common variants. Strikingly, these patterns disappear when we focus on only those variants in which the major alleles are derived. These analyses indicate that the majority of the genetic variation in terms of phenotypic consequence may result from a mutation-selection balance, as opposed to balancing selection, and have direct relevance to the study of human disease.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
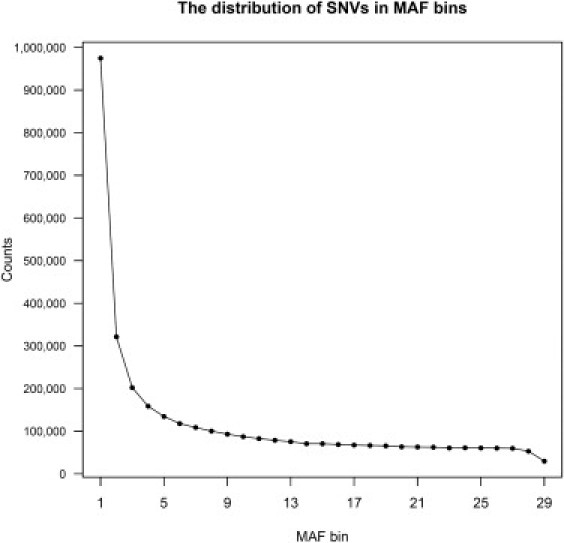
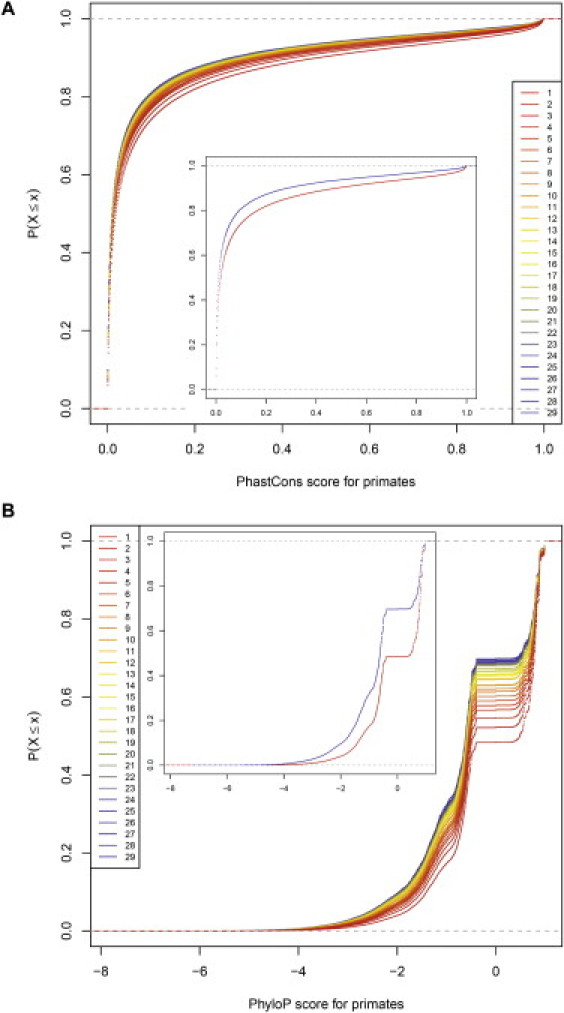
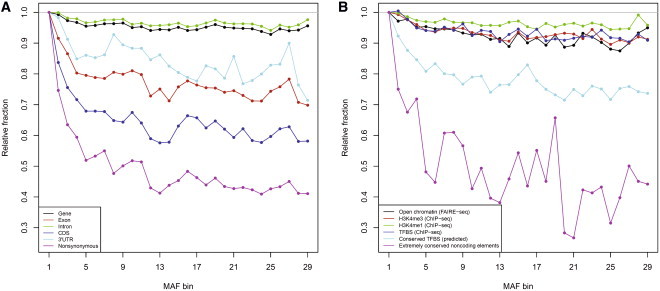
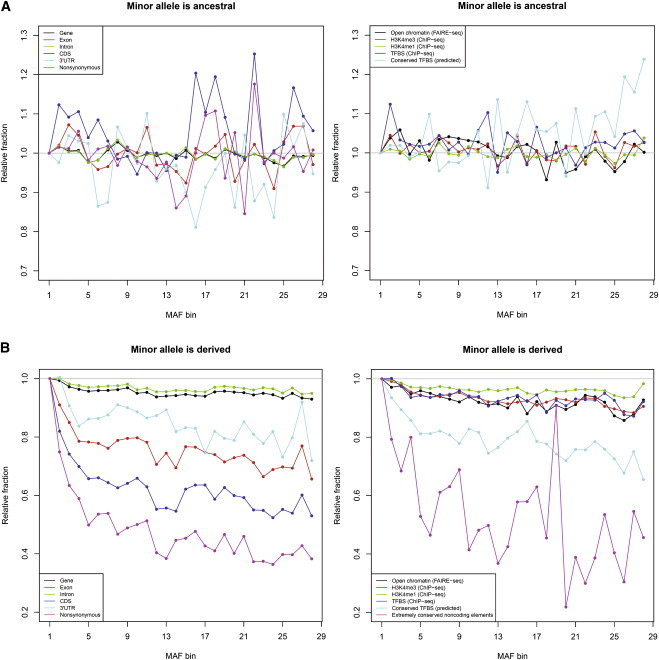
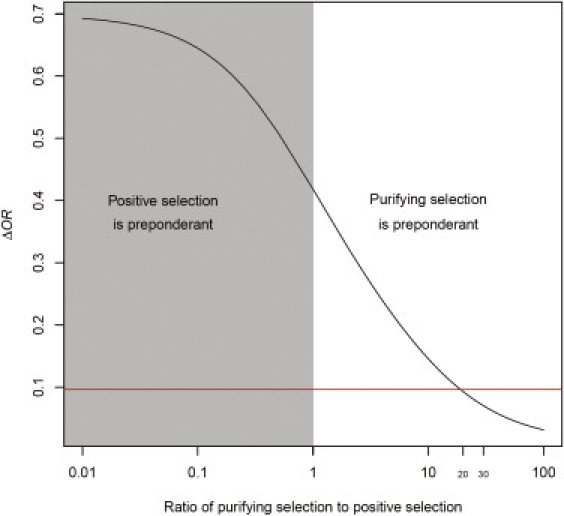
Similar articles
-
Natural Selection and Functional Potentials of Human Noncoding Elements Revealed by Analysis of Next Generation Sequencing Data.PLoS One. 2015 Jun 8;10(6):e0129023. doi: 10.1371/journal.pone.0129023. eCollection 2015. PLoS One. 2015. PMID: 26053627 Free PMC article.
-
Conserved noncoding sequences are selectively constrained and not mutation cold spots.Nat Genet. 2006 Feb;38(2):223-7. doi: 10.1038/ng1710. Epub 2005 Dec 25. Nat Genet. 2006. PMID: 16380714
-
Functional and Structural Consequence of Rare Exonic Single Nucleotide Polymorphisms: One Story, Two Tales.Genome Biol Evol. 2015 Oct 9;7(10):2929-40. doi: 10.1093/gbe/evv191. Genome Biol Evol. 2015. PMID: 26454016 Free PMC article.
-
Genome-wide scans for loci under selection in humans.Hum Genomics. 2005 Jun;2(2):113-25. doi: 10.1186/1479-7364-2-2-113. Hum Genomics. 2005. PMID: 16004726 Free PMC article. Review.
-
Common vs. rare allele hypotheses for complex diseases.Curr Opin Genet Dev. 2009 Jun;19(3):212-9. doi: 10.1016/j.gde.2009.04.010. Epub 2009 May 28. Curr Opin Genet Dev. 2009. PMID: 19481926 Free PMC article. Review.
Cited by
-
Challenges imposed by minor reference alleles on the identification and reporting of clinical variants from exome data.BMC Genomics. 2018 Jan 15;19(1):46. doi: 10.1186/s12864-018-4433-3. BMC Genomics. 2018. PMID: 29334895 Free PMC article.
-
Evidence of inbreeding depression on human height.PLoS Genet. 2012;8(7):e1002655. doi: 10.1371/journal.pgen.1002655. Epub 2012 Jul 19. PLoS Genet. 2012. PMID: 22829771 Free PMC article.
-
Genome-wide rare variant analysis for thousands of phenotypes in over 70,000 exomes from two cohorts.Nat Commun. 2020 Jan 28;11(1):542. doi: 10.1038/s41467-020-14288-y. Nat Commun. 2020. PMID: 31992710 Free PMC article.
-
Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals.Genome Res. 2014 Jan;24(1):14-24. doi: 10.1101/gr.155192.113. Epub 2013 Oct 3. Genome Res. 2014. PMID: 24092820 Free PMC article.
-
Biochemical characterization of eight genetic variants of human DNA polymerase κ involved in error-free bypass across bulky N(2)-guanyl DNA adducts.Chem Res Toxicol. 2014 May 19;27(5):919-30. doi: 10.1021/tx500072m. Epub 2014 Apr 21. Chem Res Toxicol. 2014. PMID: 24725253 Free PMC article.
References
-
- Cirulli E.T., Goldstein D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010;11:415–425. - PubMed
-
- McClellan J., King M.C. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. - PubMed
-
- Alexander R.P., Fang G., Rozowsky J., Snyder M., Gerstein M.B. Annotating non-coding regions of the genome. Nat. Rev. Genet. 2010;11:559–571. - PubMed
-
- Birney E., Stamatoyannopoulos J.A., Dutta A., Guigó R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., ENCODE Project Consortium. NISC Comparative Sequencing Program. Baylor College of Medicine Human Genome Sequencing Center. Washington University Genome Sequencing Center. Broad Institute. Children's Hospital Oakland Research Institute Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
