

Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel
- PMID: 21457909
- PMCID: PMC3071923
- DOI: 10.1016/j.ajhg.2011.03.004
Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel
Abstract
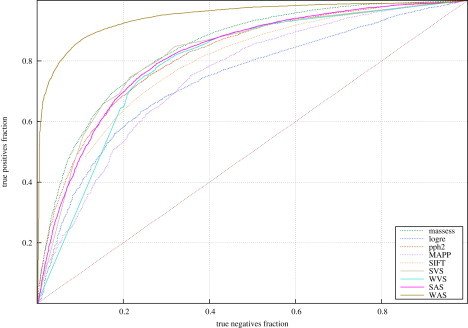
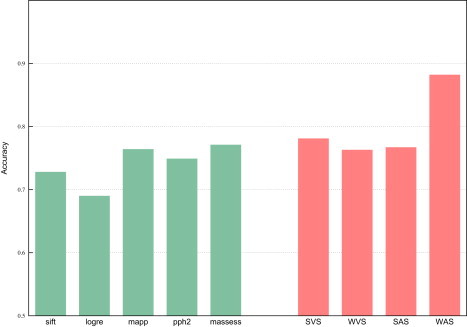
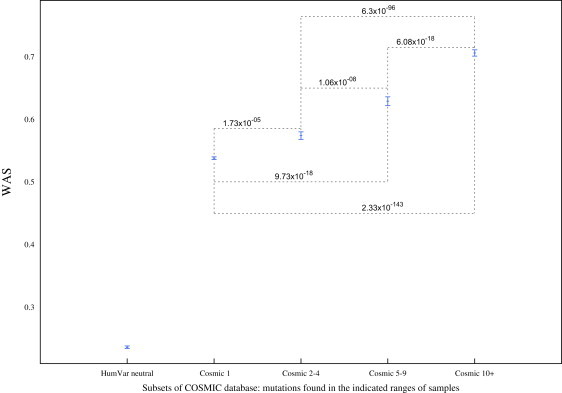
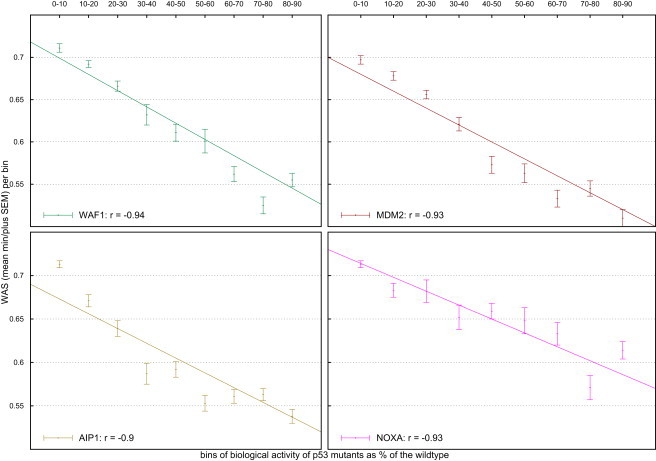
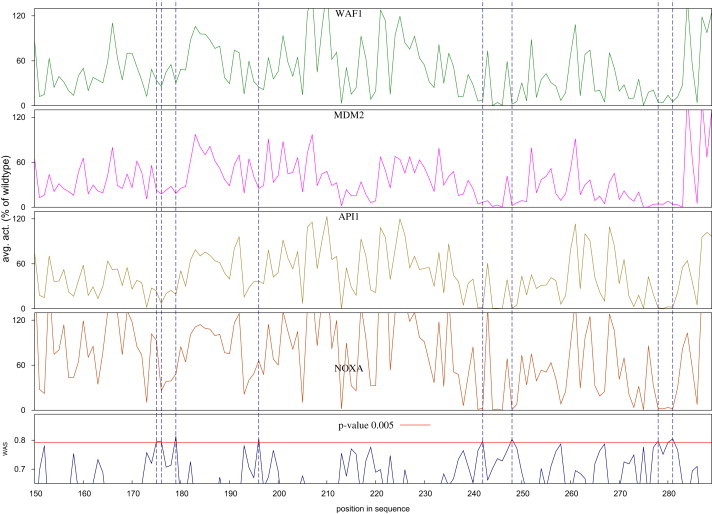
Several large ongoing initiatives that profit from next-generation sequencing technologies have driven--and in coming years will continue to drive--the emergence of long catalogs of missense single-nucleotide variants (SNVs) in the human genome. As a consequence, researchers have developed various methods and their related computational tools to classify these missense SNVs as probably deleterious or probably neutral polymorphisms. The outputs produced by each of these computational tools are of different natures and thus difficult to compare and integrate. Taking advantage of the possible complementarity between different tools might allow more accurate classifications. Here we propose an effective approach to integrating the output of some of these tools into a unified classification; this approach is based on a weighted average of the normalized scores of the individual methods (WAS). (In this paper, the approach is illustrated for the integration of five tools.) We show that this WAS outperforms each individual method in the task of classifying missense SNVs as deleterious or neutral. Furthermore, we demonstrate that this WAS can be used not only for classification purposes (deleterious versus neutral mutation) but also as an indicator of the impact of the mutation on the functionality of the mutant protein. In other words, it may be used as a deleteriousness score of missense SNVs. Therefore, we recommend the use of this WAS as a consensus deleteriousness score of missense mutations (Condel).
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
