

A de novo protein binding pair by computational design and directed evolution
- PMID: 21458342
- PMCID: PMC3102007
- DOI: 10.1016/j.molcel.2011.03.010
A de novo protein binding pair by computational design and directed evolution
Abstract
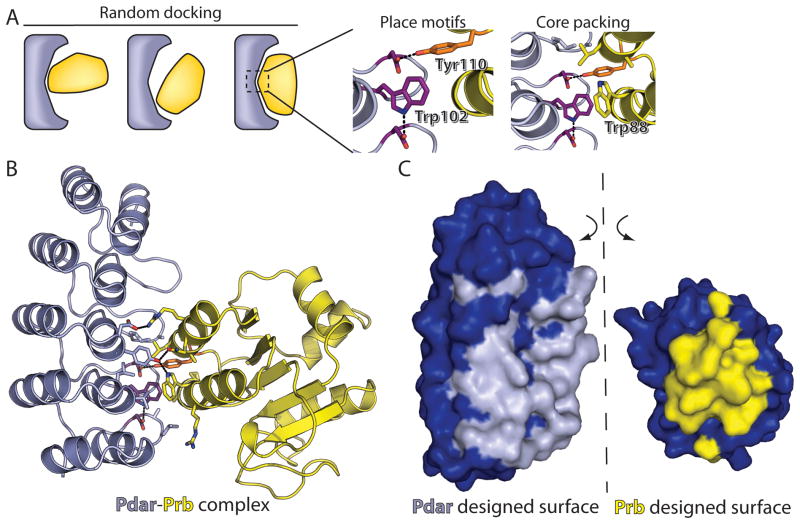
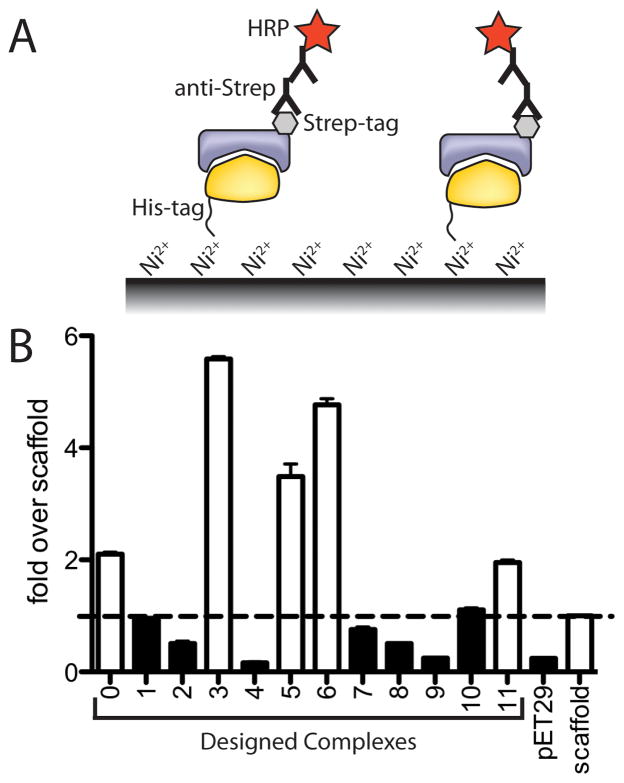
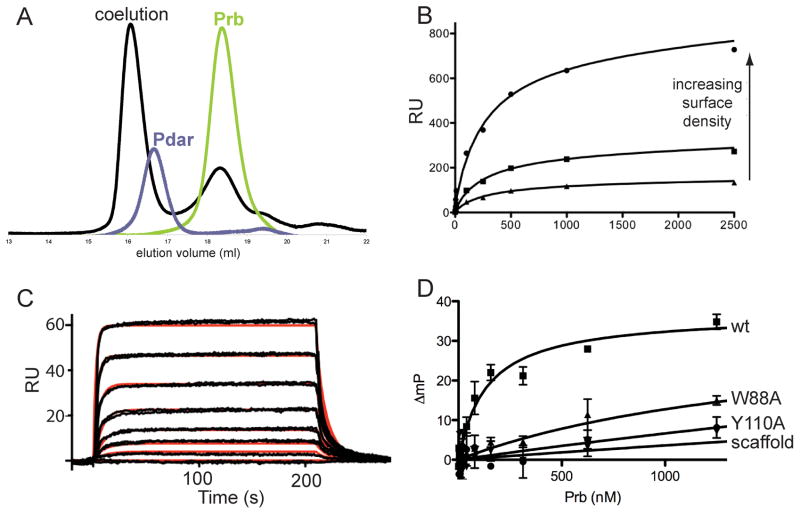
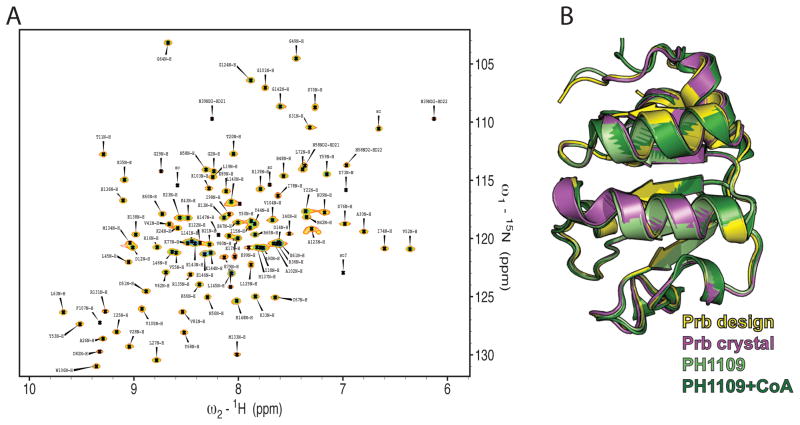
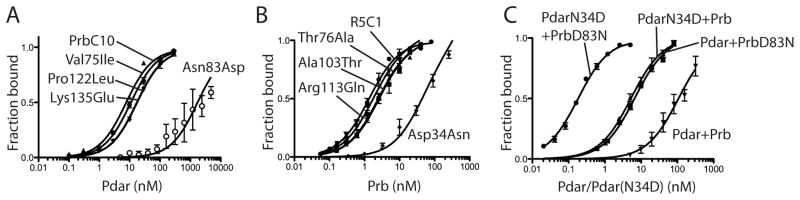
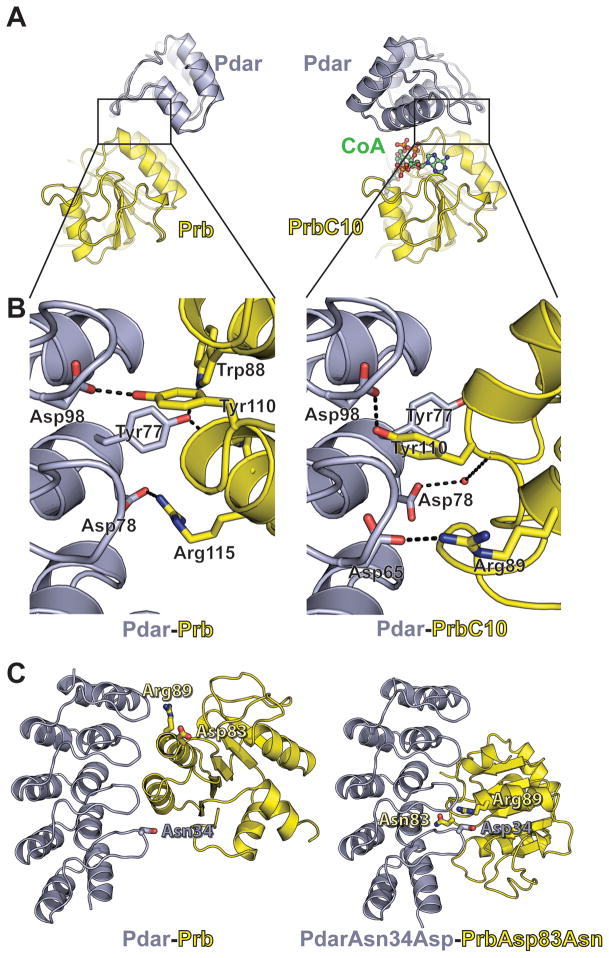
The de novo design of protein-protein interfaces is a stringent test of our understanding of the principles underlying protein-protein interactions and would enable unique approaches to biological and medical challenges. Here we describe a motif-based method to computationally design protein-protein complexes with native-like interface composition and interaction density. Using this method we designed a pair of proteins, Prb and Pdar, that heterodimerize with a Kd of 130 nM, 1000-fold tighter than any previously designed de novo protein-protein complex. Directed evolution identified two point mutations that improve affinity to 180 pM. Crystal structures of an affinity-matured complex reveal binding is entirely through the designed interface residues. Surprisingly, in the in vitro evolved complex one of the partners is rotated 180° relative to the original design model, yet still maintains the central computationally designed hotspot interaction and preserves the character of many peripheral interactions. This work demonstrates that high-affinity protein interfaces can be created by designing complementary interaction surfaces on two noninteracting partners and underscores remaining challenges.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Aharoni A, Gaidukov L, Khersonsky O, Mc QGS, Roodveldt C, Tawfik DS. The ‘evolvability’ of promiscuous protein functions. Nat Genet. 2005;37:73–76. - PubMed
-
- Aloy P, Ceulemans H, Stark A, Russell RB. The relationship between sequence and interaction divergence in proteins. J Mol Biol. 2003;332:989–998. - PubMed
-
- Batchelor AH, Piper DE, de la Brousse FC, McKnight SL, Wolberger C. The structure of GABPalpha/beta: an ETS domain- ankyrin repeat heterodimer bound to DNA. Science. 1998;279:1037–1041. - PubMed
-
- Binz HK, Amstutz P, Pluckthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–1268. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
