

Natural genetic variation caused by small insertions and deletions in the human genome
- PMID: 21460062
- PMCID: PMC3106316
- DOI: 10.1101/gr.115907.110
Natural genetic variation caused by small insertions and deletions in the human genome
Abstract
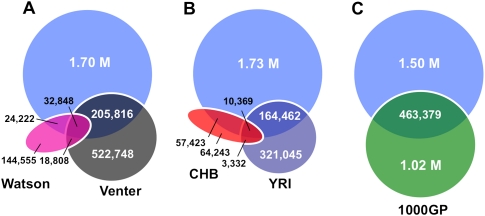
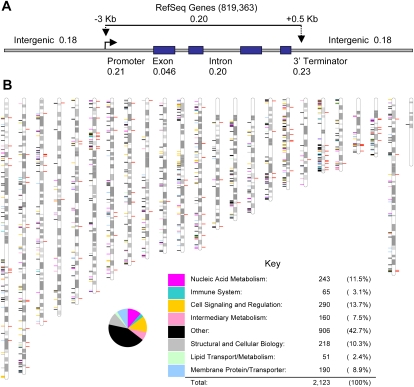
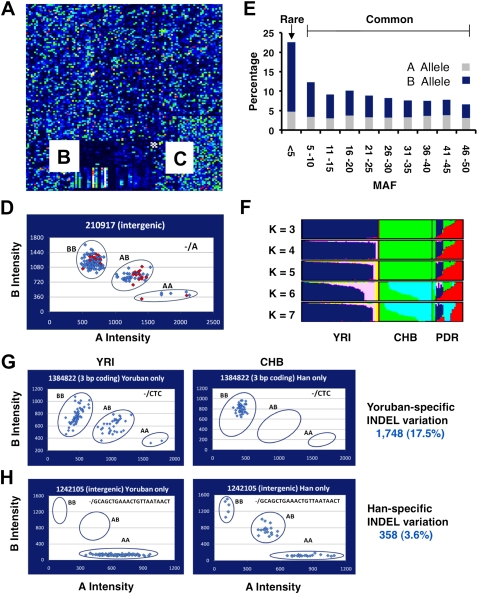
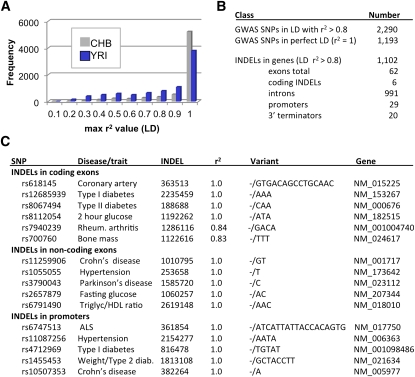
Human genetic variation is expected to play a central role in personalized medicine. Yet only a fraction of the natural genetic variation that is harbored by humans has been discovered to date. Here we report almost 2 million small insertions and deletions (INDELs) that range from 1 bp to 10,000 bp in length in the genomes of 79 diverse humans. These variants include 819,363 small INDELs that map to human genes. Small INDELs frequently were found in the coding exons of these genes, and several lines of evidence indicate that such variation is a major determinant of human biological diversity. Microarray-based genotyping experiments revealed several interesting observations regarding the population genetics of small INDEL variation. For example, we found that many of our INDELs had high levels of linkage disequilibrium (LD) with both HapMap SNPs and with high-scoring SNPs from genome-wide association studies. Overall, our study indicates that small INDEL variation is likely to be a key factor underlying inherited traits and diseases in humans.
Figures
References
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials