

Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads
- PMID: 21460107
- PMCID: PMC3127616
- DOI: 10.1128/AEM.02772-10
Generation of multimillion-sequence 16S rRNA gene libraries from complex microbial communities by assembling paired-end illumina reads
Erratum in
- Appl Environ Microbiol. 2011 Aug;77(15):5569
Abstract
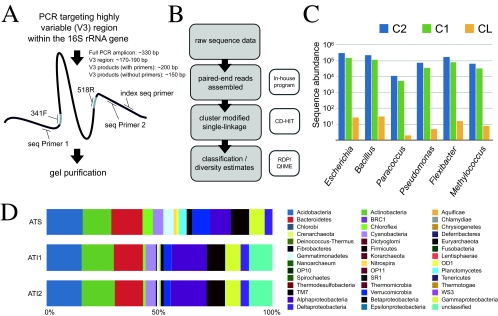
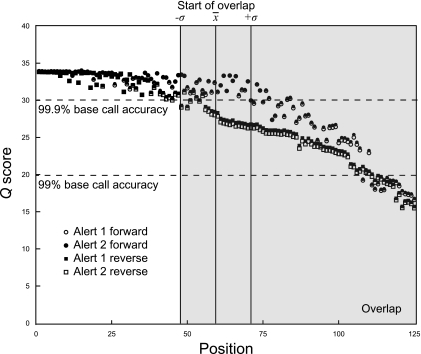
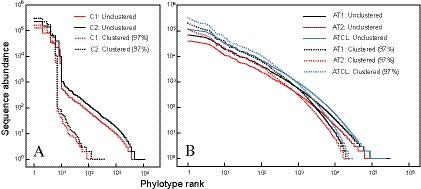
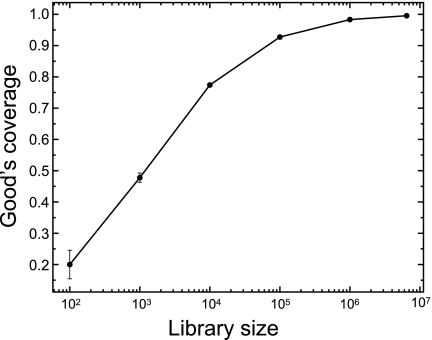
Microbial communities host unparalleled taxonomic diversity. Adequate characterization of environmental and host-associated samples remains a challenge for microbiologists, despite the advent of 16S rRNA gene sequencing. In order to increase the depth of sampling for diverse bacterial communities, we developed a method for sequencing and assembling millions of paired-end reads from the 16S rRNA gene (spanning the V3 region; ∼200 nucleotides) by using an Illumina genome analyzer. To confirm reproducibility and to identify a suitable computational pipeline for data analysis, sequence libraries were prepared in duplicate for both a defined mixture of DNAs from known cultured bacterial isolates (>1 million postassembly sequences) and an Arctic tundra soil sample (>6 million postassembly sequences). The Illumina 16S rRNA gene libraries represent a substantial increase in number of sequences over all extant next-generation sequencing approaches (e.g., 454 pyrosequencing), while the assembly of paired-end 125-base reads offers a methodological advantage by incorporating an initial quality control step for each 16S rRNA gene sequence. This method incorporates indexed primers to enable the characterization of multiple microbial communities in a single flow cell lane, may be modified readily to target other variable regions or genes, and demonstrates unprecedented and economical access to DNAs from organisms that exist at low relative abundances.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
