

Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome
- PMID: 21464306
- PMCID: PMC3081023
- DOI: 10.1073/pnas.1103471108
Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome
Abstract
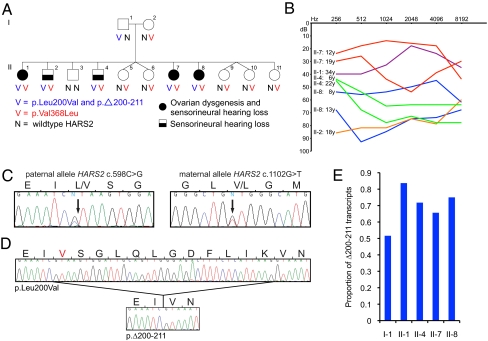
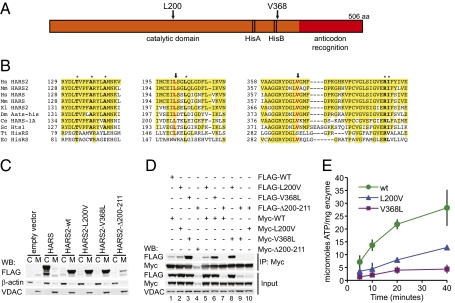
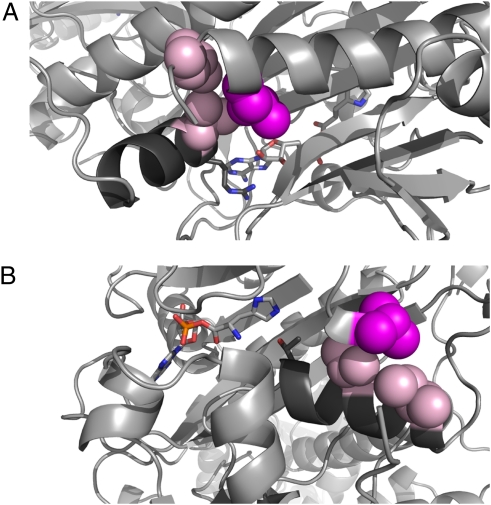
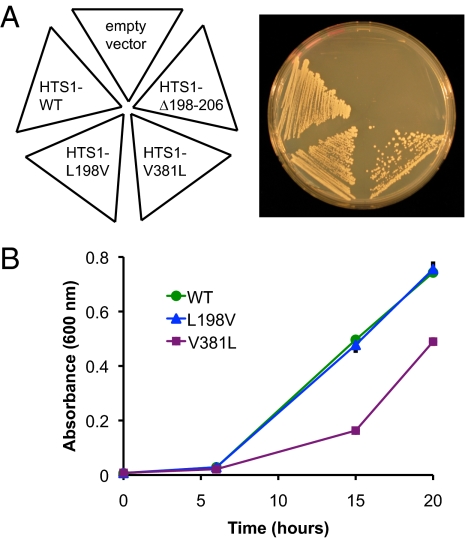
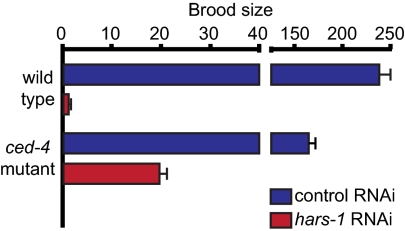
Perrault syndrome is a genetically heterogeneous recessive disorder characterized by ovarian dysgenesis and sensorineural hearing loss. In a nonconsanguineous family with five affected siblings, linkage analysis and genomic sequencing revealed the genetic basis of Perrault syndrome to be compound heterozygosity for mutations in the mitochondrial histidyl tRNA synthetase HARS2 at two highly conserved amino acids, L200V and V368L. The nucleotide substitution creating HARS2 p.L200V also created an alternate splice leading to deletion of 12 codons from the HARS2 message. Affected family members thus carried three mutant HARS2 transcripts. Aminoacylation activity of HARS2 p.V368L and HARS2 p.L200V was reduced and the deletion mutant was not stably expressed in mammalian mitochondria. In yeast, lethality of deletion of the single essential histydyl tRNA synthetase HTS1 was fully rescued by wild-type HTS1 and by HTS1 p.L198V (orthologous to HARS2 p.L200V), partially rescued by HTS1 p.V381L (orthologous to HARS2 p.V368L), and not rescued by the deletion mutant. In Caenorhabditis elegans, reduced expression by RNAi of the single essential histydyl tRNA synthetase hars-1 severely compromised fertility. Together, these data suggest that Perrault syndrome in this family was caused by reduction of HARS2 activity. These results implicate aberrations of mitochondrial translation in mammalian gonadal dysgenesis. More generally, the relationship between HARS2 and Perrault syndrome illustrates how causality may be demonstrated for extremely rare inherited mutations in essential, highly conserved genes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Perrault M, Klotz B, Housset E. [Two cases of Turner syndrome with deaf-mutism in two sisters] Bull Mem Soc Med Hop Paris. 1951;67:79–84. - PubMed
-
- Christakos AC, Simpson JL, Younger JB, Christian CD. Gonadal dysgenesis as an autosomal recessive condition. Am J Obstet Gynecol. 1969;104:1027–1030. - PubMed
-
- Amor DJ, Delatycki MB, Gardner RJ, Storey E. New variant of familial cerebellar ataxia with hypergonadotropic hypogonadism and sensorineural deafness. Am J Med Genet. 2001;99:29–33. - PubMed
-
- Marlin S, et al. Perrault syndrome: Report of four new cases, review and exclusion of candidate genes. Am J Med Genet A. 2008;146A:661–664. - PubMed
-
- Kobe C, Kracht LW, Timmermann L, Bachmann J, Schmidt MC. Perrault syndrome with progressive nervous system involvement. Clin Nucl Med. 2008;33:922–924. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
