

Intracellular leptin-signaling pathways in hypothalamic neurons: the emerging role of phosphatidylinositol-3 kinase-phosphodiesterase-3B-cAMP pathway
- PMID: 21464566
- PMCID: PMC3130491
- DOI: 10.1159/000326785
Intracellular leptin-signaling pathways in hypothalamic neurons: the emerging role of phosphatidylinositol-3 kinase-phosphodiesterase-3B-cAMP pathway
Abstract
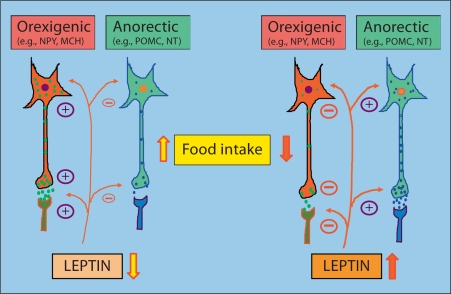
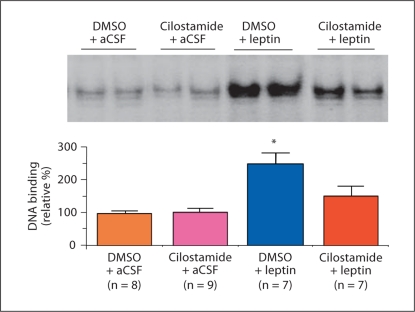
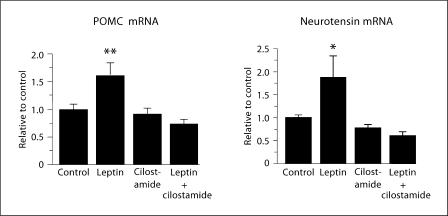
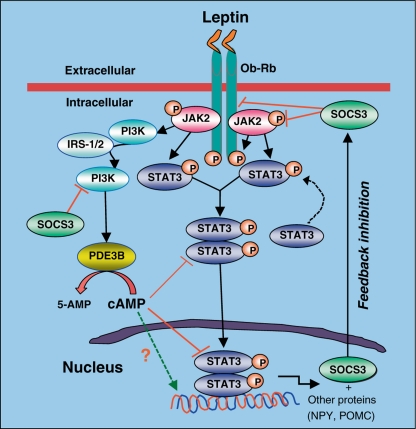
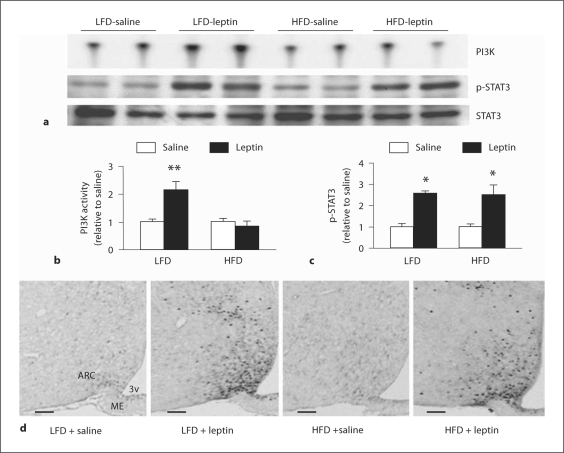
Leptin is secreted primarily by fat cells and acts centrally, particularly in the hypothalamus, to reduce food intake and body weight. Besides the classical JAK2 (Janus kinase-2)-STAT3 (signal transducer and activator of transcription-3) pathway, several non-STAT3 pathways play an important role in mediating leptin signaling in the hypothalamus. We have demonstrated that leptin action in the hypothalamus is mediated by an insulin-like signaling pathway involving stimulation of PI3K (phosphatidylinositol-3 kinase) and PDE3B (phosphodiesterase-3B), and reduction in cAMP levels, and that a PI3K-PDE3B-cAMP pathway interacting with the JAK2-STAT3 pathway constitutes a critical component of leptin signaling in the hypothalamus. It appears that defective regulation of multiple signaling pathways in the hypothalamus causes central leptin resistance, a major cause of obesity. In this regard, we have shown that leptin resistance in hypothalamic neurons following chronic central infusion of this hormone is associated with a defect in the PI3K-PDE3B-cAMP, and not due to compromised signaling in the JAK2-STAT3 pathway. Similarly, the PI3K, but not the STAT3, pathway is impaired in the hypothalamus during the development of diet-induced obesity. Additionally, our recent work suggests that suppressor of cytokine signaling-3 negatively regulates the PI3K pathway of leptin signaling in the hypothalamus, a mechanism expected to play a significant role in diet-induced obesity. Together, the PI3K-PDE3B-cAMP pathway appears to emerge as a major mechanism of leptin signaling in the hypothalamus in regulating energy balance.
Copyright © 2011 S. Karger AG, Basel.
Figures
Similar articles
-
Phosphatidylinositol 3-kinase is an upstream regulator of the phosphodiesterase 3B pathway of leptin signalling that may not involve activation of Akt in the rat hypothalamus.J Neuroendocrinol. 2013 Feb;25(2):168-79. doi: 10.1111/j.1365-2826.2012.02386.x. J Neuroendocrinol. 2013. PMID: 22967108 Free PMC article.
-
Hypothalamic phosphatidylinositol 3-kinase-phosphodiesterase 3B-cyclic AMP pathway of leptin signalling is impaired following chronic central leptin infusion.J Neuroendocrinol. 2005 Nov;17(11):720-6. doi: 10.1111/j.1365-2826.2005.01362.x. J Neuroendocrinol. 2005. PMID: 16219000
-
Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance.Front Neuroendocrinol. 2003 Dec;24(4):225-53. doi: 10.1016/j.yfrne.2003.10.001. Front Neuroendocrinol. 2003. PMID: 14726256 Review.
-
A phosphatidylinositol 3-kinase phosphodiesterase 3B-cyclic AMP pathway in hypothalamic action of leptin on feeding.Nat Neurosci. 2002 Aug;5(8):727-8. doi: 10.1038/nn885. Nat Neurosci. 2002. PMID: 12101402
-
Minireview: A hypothalamic role in energy balance with special emphasis on leptin.Endocrinology. 2004 Jun;145(6):2613-20. doi: 10.1210/en.2004-0032. Epub 2004 Mar 24. Endocrinology. 2004. PMID: 15044360 Review.
Cited by
-
Phosphatidylinositol 3-kinase is an upstream regulator of the phosphodiesterase 3B pathway of leptin signalling that may not involve activation of Akt in the rat hypothalamus.J Neuroendocrinol. 2013 Feb;25(2):168-79. doi: 10.1111/j.1365-2826.2012.02386.x. J Neuroendocrinol. 2013. PMID: 22967108 Free PMC article.
-
Leptin Signaling in the Control of Metabolism and Appetite: Lessons from Animal Models.J Mol Neurosci. 2018 Nov;66(3):390-402. doi: 10.1007/s12031-018-1185-0. Epub 2018 Oct 3. J Mol Neurosci. 2018. PMID: 30284225 Review.
-
Hypothalamic Phosphodiesterase 3B Pathway Mediates Anorectic and Body Weight-Reducing Effects of Insulin in Male Mice.Neuroendocrinology. 2017;104(2):145-156. doi: 10.1159/000445523. Epub 2016 Mar 23. Neuroendocrinology. 2017. PMID: 27002827 Free PMC article.
-
A novel thermoregulatory role for PDE10A in mouse and human adipocytes.EMBO Mol Med. 2016 Jul 1;8(7):796-812. doi: 10.15252/emmm.201506085. Print 2016 Jul. EMBO Mol Med. 2016. PMID: 27247380 Free PMC article.
-
Energy homeostasis deregulation is attenuated by TUDCA treatment in streptozotocin-induced Alzheimer's disease mice model.Sci Rep. 2021 Sep 13;11(1):18114. doi: 10.1038/s41598-021-97624-6. Sci Rep. 2021. PMID: 34518585 Free PMC article.
References
-
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. - PubMed
-
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. - PubMed
-
- Sahu A. Leptin signaling in the hypothalamus: emphasis on energy homeostasis and leptin resistance. Front Neuroendocrinol. 2003;24:225–253. - PubMed
-
- Yadav VK, Oury F, Suda N, Liu ZW, Gao XB, Confavreux C, Klemenhagen KC, Tanaka KF, Gingrich JA, Guo XE, Tecott LH, Mann JJ, Hen R, Horvath TL, Karsenty G. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138:976–989. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
