

Synergistic interactions between heregulin and peroxisome proliferator-activated receptor-gamma (PPARgamma) agonist in breast cancer cells
- PMID: 21467033
- PMCID: PMC3103381
- DOI: 10.1074/jbc.M110.191718
Synergistic interactions between heregulin and peroxisome proliferator-activated receptor-gamma (PPARgamma) agonist in breast cancer cells
Abstract
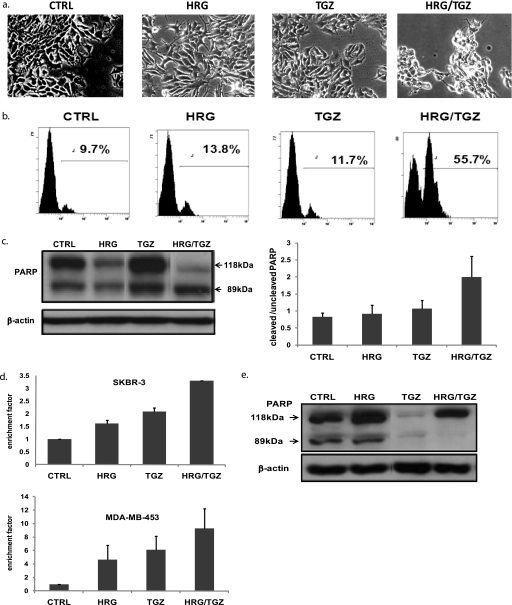
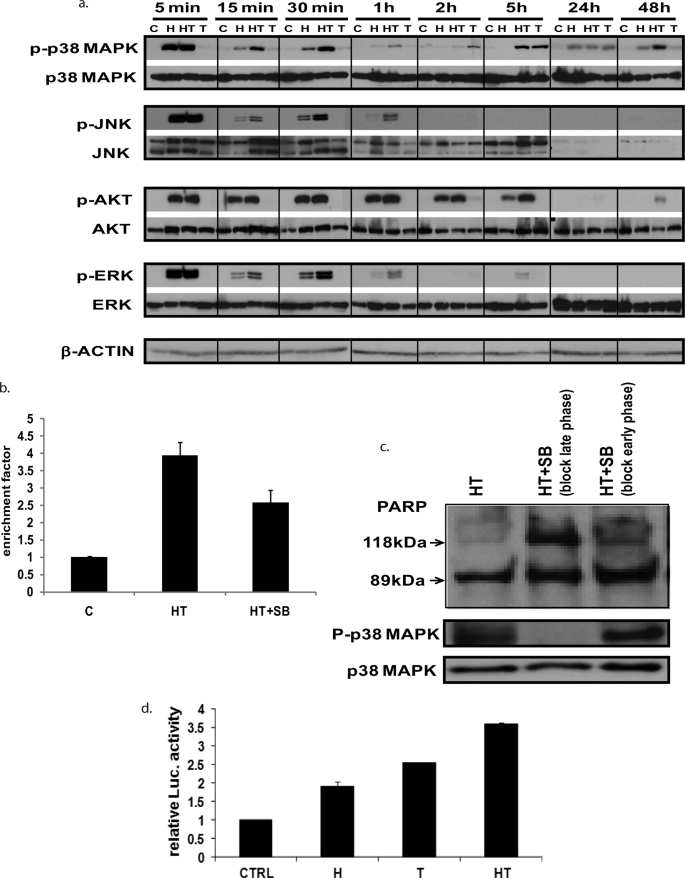
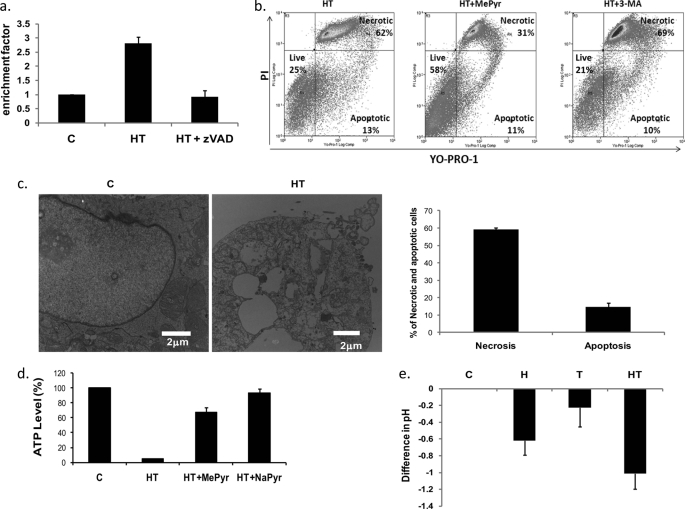
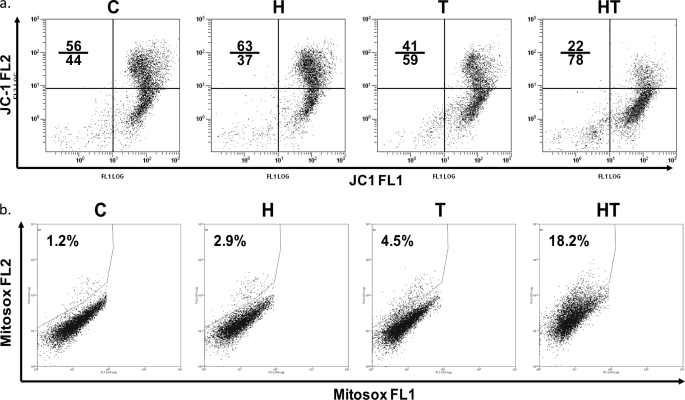
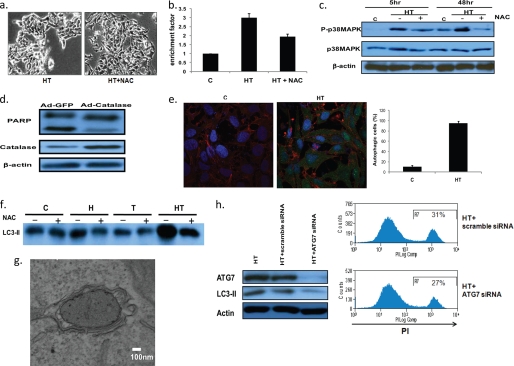
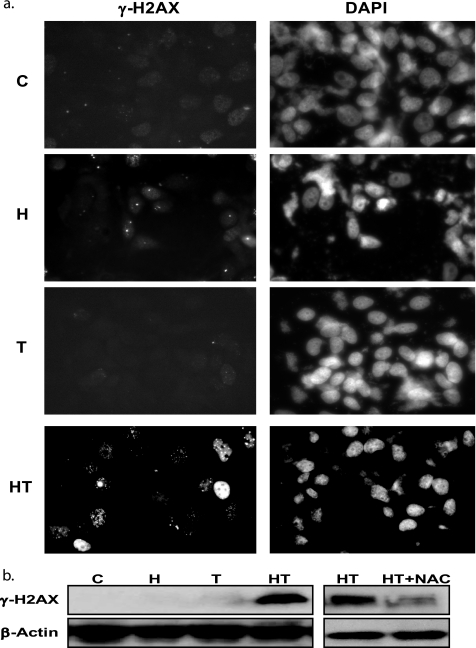
Here, we demonstrate that troglitazone (Rezulin), a peroxisome proliferator-activated receptor agonist, acted in synergy with heregulin to induce massive cell death in breast cancer cells. Although the combination of heregulin and troglitazone (HRG/TGZ) induced both apoptosis and necrosis, the main mode of cell death was caspase-independent and occurred via necrosis. This combination increased generation of superoxide in mitochondria, which in turn destabilized mitochondria potential. Pretreatment with N-acetyl-l-cysteine and catalase expression ameliorated cell death induced by the combination treatment, indicating a role of oxidative stress in mediating HRG/TGZ-induced cell death. Notably, pretreatment with pyruvate significantly prevented the cell death, suggesting a potential mechanistic link between metabolic stress and HRG/TGZ-induced cell death. The activation of the HRG signaling axis has been considered as a poor prognostic factor in breast cancer and confers resistance to gefitinib (Iressa) and tamoxifen. However, our data presented here paradoxically suggest that HRG expression can actually be beneficial when it comes to treating breast cancer with peroxisome proliferator-activated receptor-γ ligands. Taken together, the combination of HRG and TGZ may provide a basis for the development of a novel strategy in the treatment of apoptosis-resistant and/or hormone-refractory breast cancer.
Figures
Similar articles
-
Inhibition of ERK activity enhances the cytotoxic effect of peroxisome proliferator-activated receptor γ (PPARγ) agonists in HeLa cells.Biochem Biophys Res Commun. 2017 Jan 22;482(4):843-848. doi: 10.1016/j.bbrc.2016.11.122. Epub 2016 Nov 23. Biochem Biophys Res Commun. 2017. PMID: 27888104
-
PPARγ-inactive Δ2-troglitazone independently triggers ER stress and apoptosis in breast cancer cells.Mol Carcinog. 2015 May;54(5):393-404. doi: 10.1002/mc.22109. Epub 2013 Nov 30. Mol Carcinog. 2015. PMID: 24293218
-
New troglitazone derivatives devoid of PPARγ agonist activity display an increased antiproliferative effect in both hormone-dependent and hormone-independent breast cancer cell lines.Breast Cancer Res Treat. 2010 Nov;124(1):101-10. doi: 10.1007/s10549-009-0700-y. Epub 2010 Jan 7. Breast Cancer Res Treat. 2010. PMID: 20054646
-
The role of peroxisome proliferator-activated receptor-γ in breast cancer.Anticancer Agents Med Chem. 2012 Nov;12(9):1025-44. doi: 10.2174/187152012803529664. Anticancer Agents Med Chem. 2012. PMID: 22583414 Review.
-
PPARgamma-independent antitumor effects of thiazolidinediones.Cancer Lett. 2009 Apr 18;276(2):119-24. doi: 10.1016/j.canlet.2008.08.008. Epub 2008 Sep 13. Cancer Lett. 2009. PMID: 18790559 Free PMC article. Review.
Cited by
-
The Key to Unlocking the Chemotherapeutic Potential of PPARγ Ligands: Having the Right Combination.PPAR Res. 2012;2012:946943. doi: 10.1155/2012/946943. Epub 2012 Jul 2. PPAR Res. 2012. PMID: 22966225 Free PMC article.
-
Pharmacological inhibition of Peroxisome Proliferation-Activated Receptor Delta (PPARδ) imparts selective leukemia cell death.Cancer Metab. 2025 Jul 25;13(1):36. doi: 10.1186/s40170-025-00402-5. Cancer Metab. 2025. PMID: 40713763 Free PMC article.
-
Chemotherapy and chemoprevention by thiazolidinediones.Biomed Res Int. 2015;2015:845340. doi: 10.1155/2015/845340. Epub 2015 Mar 19. Biomed Res Int. 2015. PMID: 25866814 Free PMC article. Review.
-
PPAR-γ Modulators as Current and Potential Cancer Treatments.Front Oncol. 2021 Sep 23;11:737776. doi: 10.3389/fonc.2021.737776. eCollection 2021. Front Oncol. 2021. PMID: 34631571 Free PMC article. Review.
-
The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention.Nat Rev Cancer. 2012 Feb 9;12(3):181-95. doi: 10.1038/nrc3214. Nat Rev Cancer. 2012. PMID: 22318237 Free PMC article. Review.
References
-
- Le X. F., Varela C. R., Bast R. C., Jr. (2002) Apoptosis 7, 483–491 - PubMed
-
- Lupu R., Cardillo M., Cho C., Harris L., Hijazi M., Perez C., Rosenberg K., Yang D., Tang C. (1996) Breast Cancer Res. Treat. 38, 57–66 - PubMed
-
- Vadlamudi R., Adam L., Tseng B., Costa L., Kumar R. (1999) Cancer Res. 59, 2843–2846 - PubMed
-
- Xu F., Yu Y., Le X. F., Boyer C., Mills G. B., Bast R. C., Jr. (1999) Clin. Cancer Res. 5, 3653–3660 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
