

Alzheimer's disease: the challenge of the second century
- PMID: 21471435
- PMCID: PMC3130546
- DOI: 10.1126/scitranslmed.3002369
Alzheimer's disease: the challenge of the second century
Abstract
Alzheimer's disease (AD) was first described a little more than 100 years ago. It is the most common cause of dementia with an estimated prevalence of 30 million people worldwide, a number that is expected to quadruple in 40 years. There currently is no effective treatment that delays the onset or slows the progression of AD. However, major scientific advances in the areas of genetics, biochemistry, cell biology, and neuroscience over the past 25 years have changed the way we think about AD. This review discusses some of the challenges to translating these basic molecular and cellular discoveries into clinical therapies. Current information suggests that if the disease is detected before the onset of overt symptoms, it is possible that treatments based on knowledge of underlying pathogenesis can and will be effective.
Figures
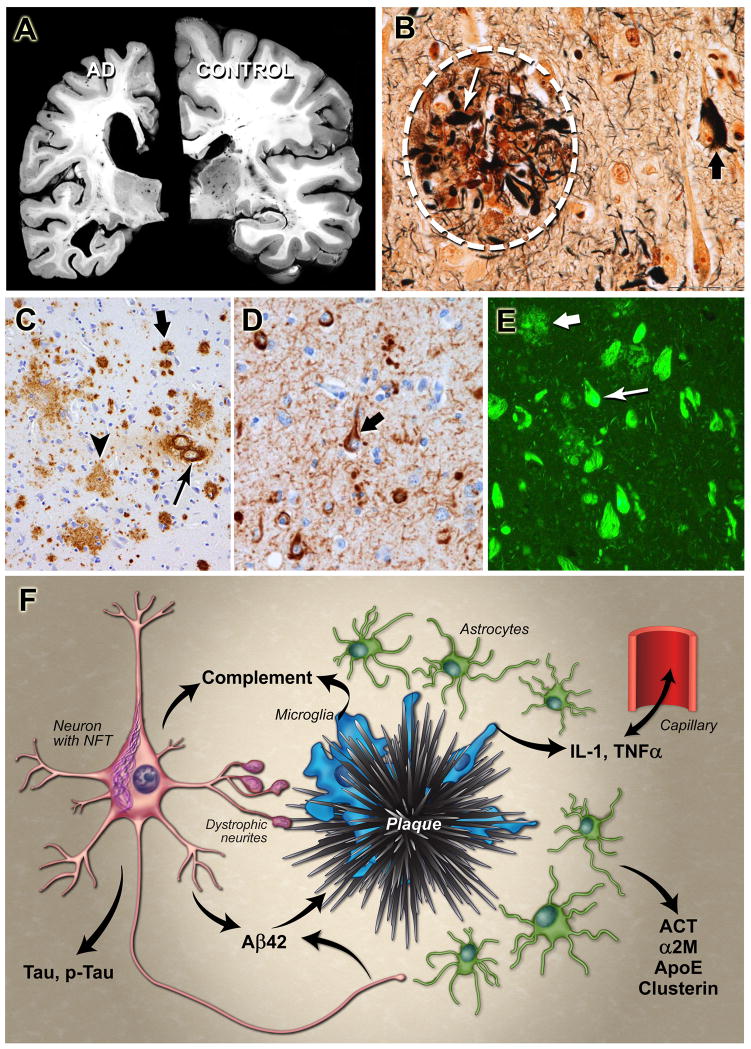
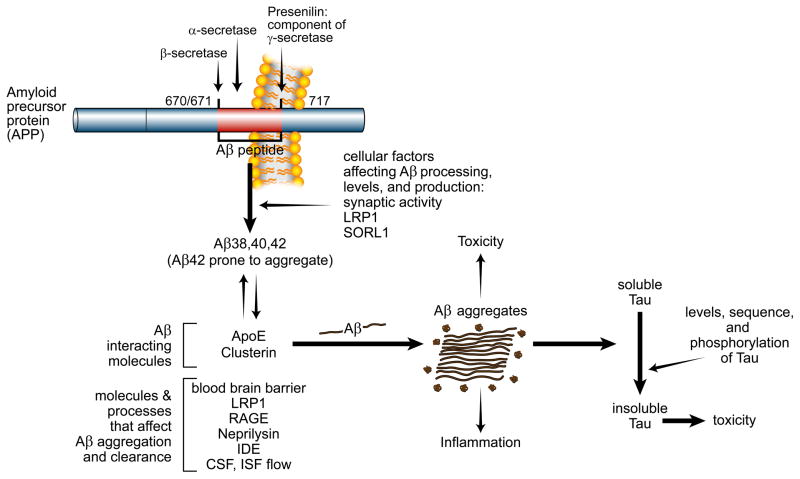
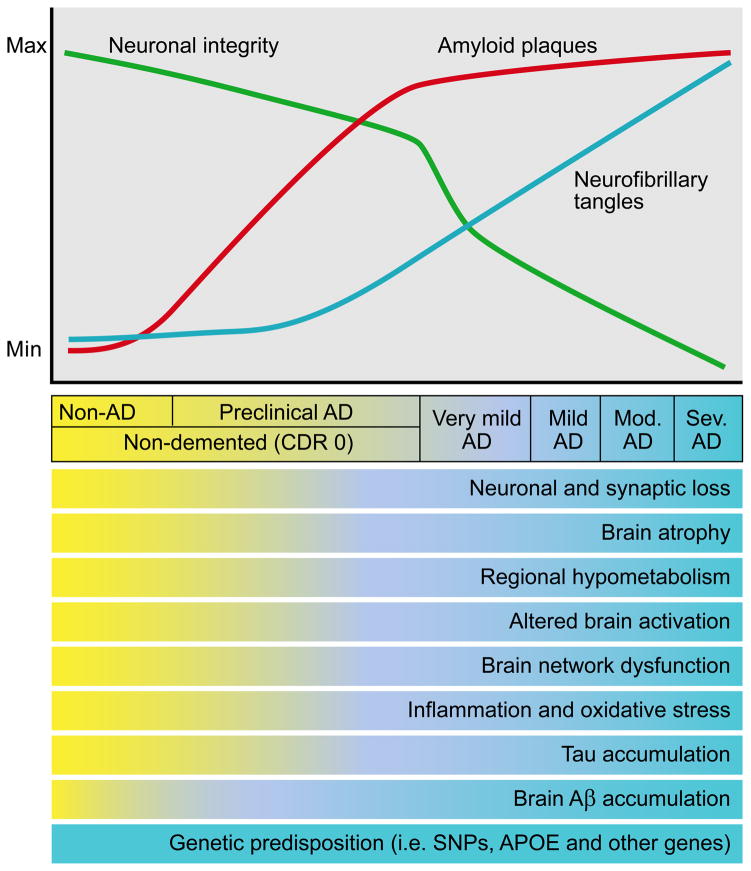
References
-
- Alzheimer A. Über eine eigenartige Erkrankung der Hirnrinde. Allg Z Psychiatr. 1907;64:146–148.
-
- Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, Waters C, Jimison P, Shepherd E, Sevush S, Graff-Radford N, Newland D, Todd M, Miller B, Gold M, Heilman K, Doty L, Goodman I, Robinson B, Pearl G, Dickson D, Duara R. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16:203–212. - PubMed
-
- Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. - PubMed
-
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
