

Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis
- PMID: 21473986
- PMCID: PMC3071922
- DOI: 10.1016/j.ajhg.2011.03.015
Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis
Abstract
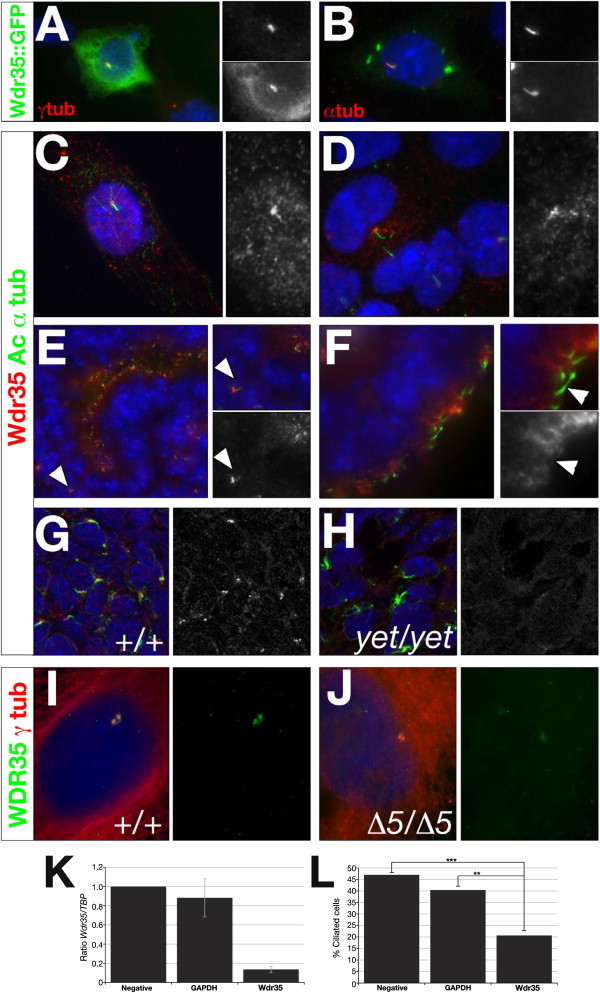
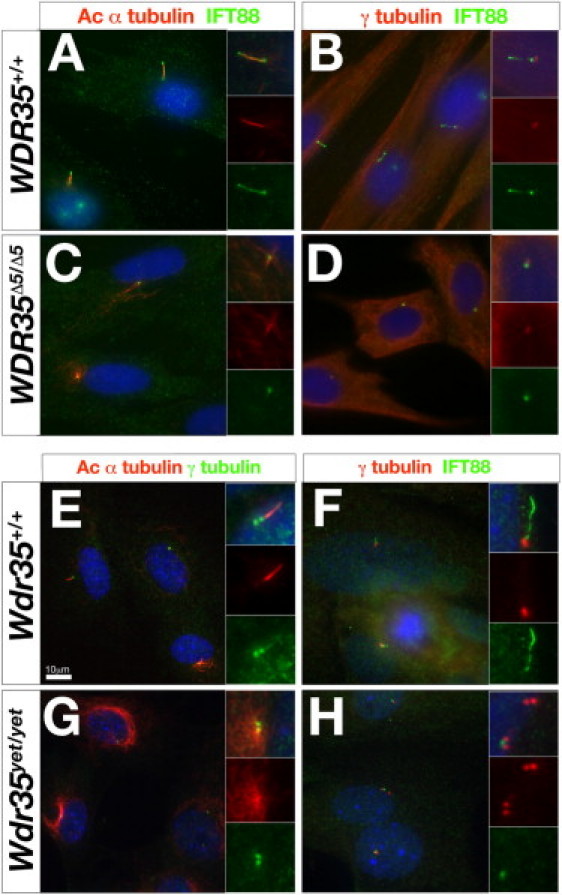
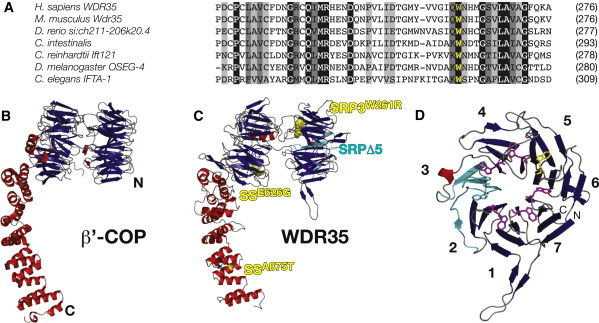
Defects in cilia formation and function result in a range of human skeletal and visceral abnormalities. Mutations in several genes have been identified to cause a proportion of these disorders, some of which display genetic (locus) heterogeneity. Mouse models are valuable for dissecting the function of these genes, as well as for more detailed analysis of the underlying developmental defects. The short-rib polydactyly (SRP) group of disorders are among the most severe human phenotypes caused by cilia dysfunction. We mapped the disease locus from two siblings affected by a severe form of SRP to 2p24, where we identified an in-frame homozygous deletion of exon 5 in WDR35. We subsequently found compound heterozygous missense and nonsense mutations in WDR35 in an independent second case with a similar, severe SRP phenotype. In a mouse mutation screen for developmental phenotypes, we identified a mutation in Wdr35 as the cause of midgestation lethality, with abnormalities characteristic of defects in the Hedgehog signaling pathway. We show that endogenous WDR35 localizes to cilia and centrosomes throughout the developing embryo and that human and mouse fibroblasts lacking the protein fail to produce cilia. Through structural modeling, we show that WDR35 has strong homology to the COPI coatamers involved in vesicular trafficking and that human SRP mutations affect key structural elements in WDR35. Our report expands, and sheds new light on, the pathogenesis of the SRP spectrum of ciliopathies.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. - PubMed
-
- Baker K., Beales P.L. Making sense of cilia in disease: the human ciliopathies. Am. J. Med. Genet. C. Semin. Med. Genet. 2009;151C:281–295. - PubMed
-
- Ho N.C., Francomano C.A., van Allen M. Jeune asphyxiating thoracic dystrophy and short-rib polydactyly type III (Verma-Naumoff) are variants of the same disorder. Am. J. Med. Genet. 2000;90:310–314. - PubMed
-
- Amar M.J., Sutphen R., Kousseff B.G. Expanded phenotype of cranioectodermal dysplasia (Sensenbrenner syndrome) Am. J. Med. Genet. 1997;70:349–352. - PubMed
-
- Pedersen L.B., Rosenbaum J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008;85:23–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
