

Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis
- PMID: 21474066
- PMCID: PMC3145340
- DOI: 10.1016/j.molcel.2011.02.020
Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis
Abstract
Mutations in both RAS and the PTEN/PIK3CA/AKT signaling module are found in the same human tumors. PIK3CA and AKT are downstream effectors of RAS, and the selective advantage conferred by mutation of two genes in the same pathway is unclear. Based on a comparative molecular analysis, we show that activated PIK3CA/AKT is a weaker inducer of senescence than is activated RAS. Moreover, concurrent activation of RAS and PIK3CA/AKT impairs RAS-induced senescence. In vivo, bypass of RAS-induced senescence by activated PIK3CA/AKT correlates with accelerated tumorigenesis. Thus, not all oncogenes are equally potent inducers of senescence, and, paradoxically, a weak inducer of senescence (PIK3CA/AKT) can be dominant over a strong inducer of senescence (RAS). For tumor growth, one selective advantage of concurrent mutation of RAS and PTEN/PIK3CA/AKT is suppression of RAS-induced senescence. Evidence is presented that this new understanding can be exploited in rational development and targeted application of prosenescence cancer therapies.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
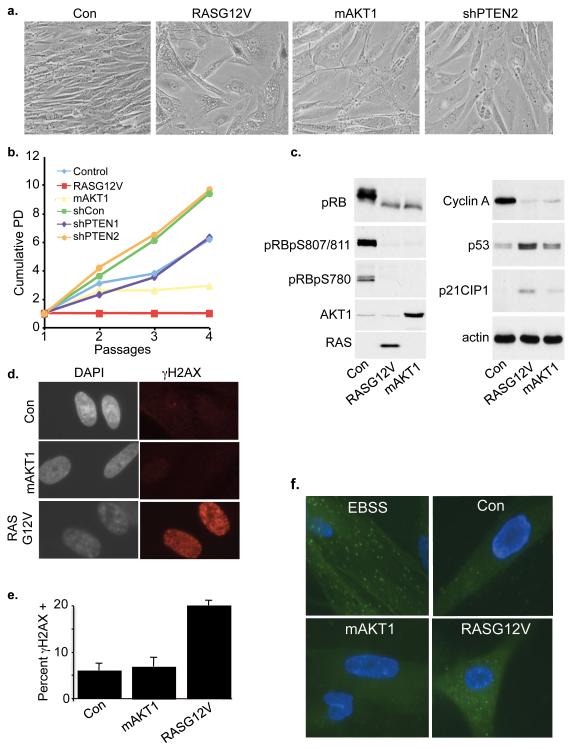
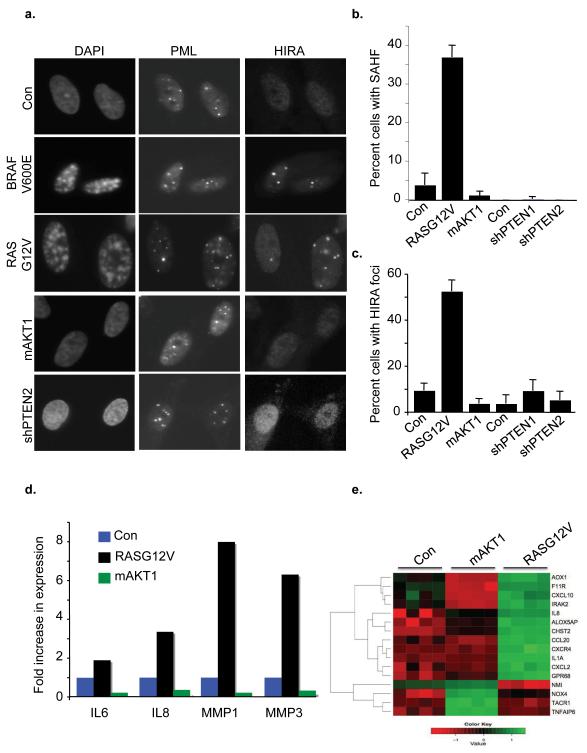
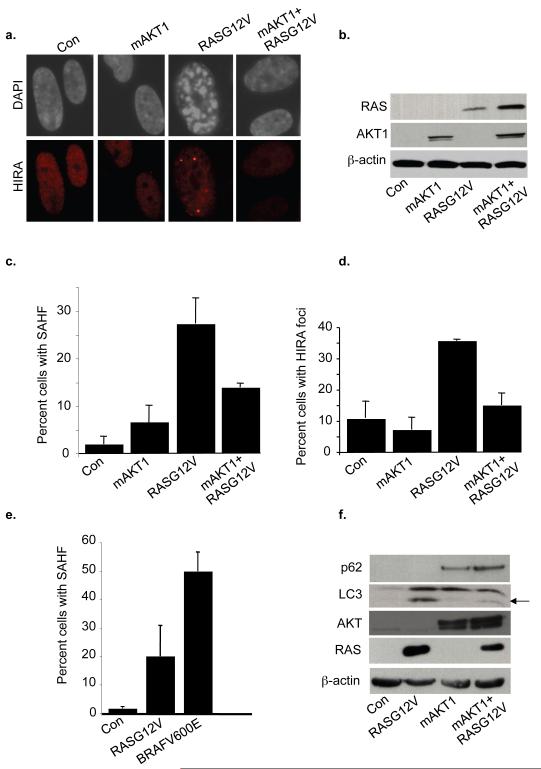
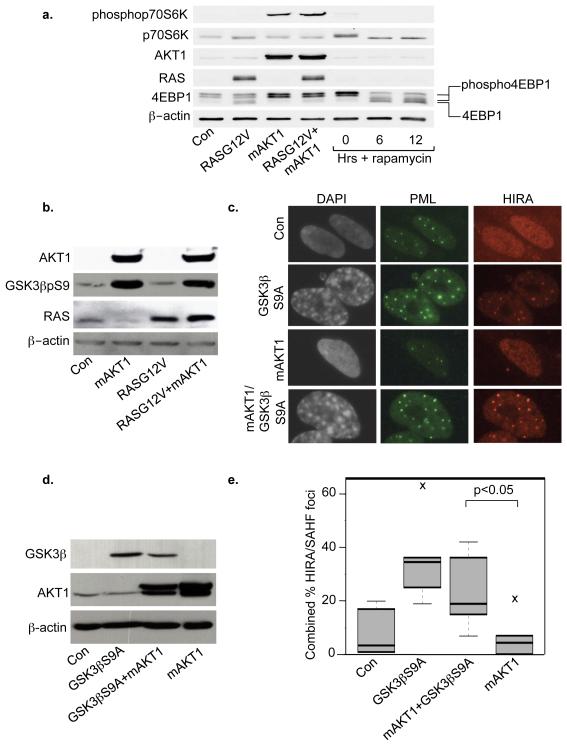
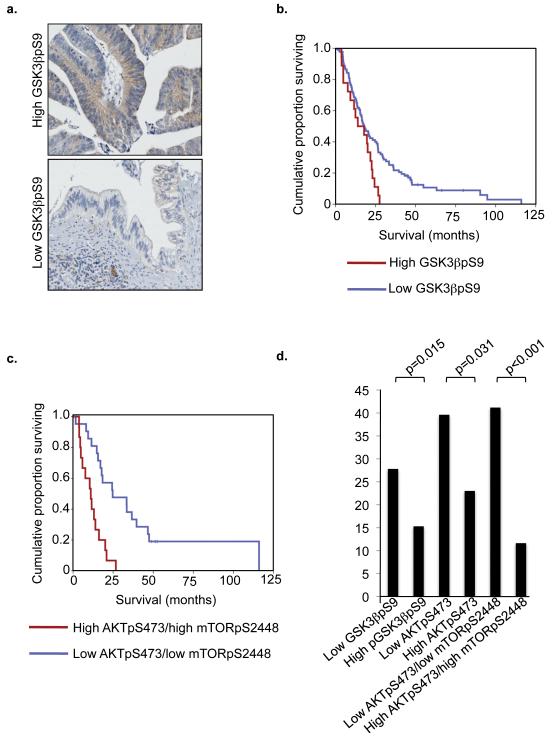
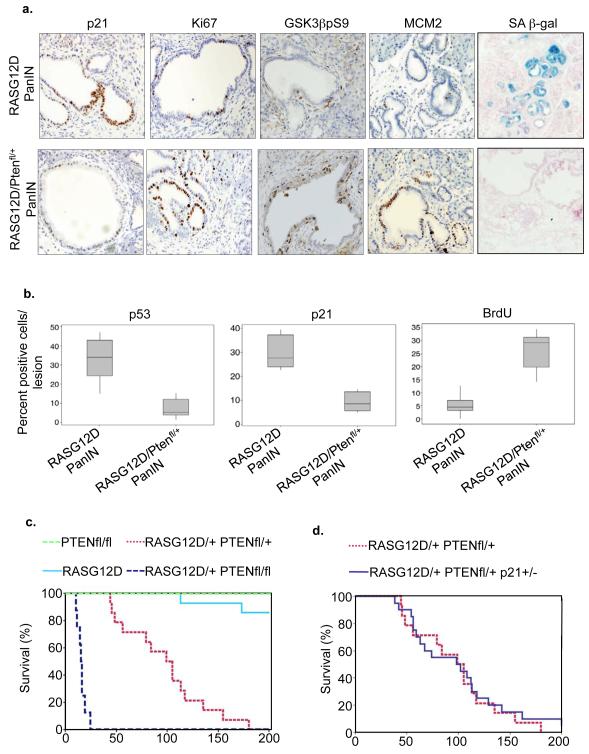
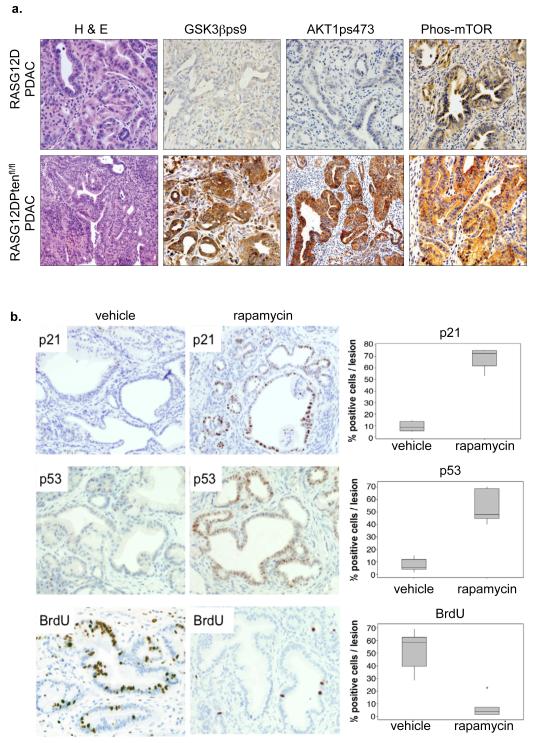
Comment in
-
Senescence: Double or quit?Nat Rev Cancer. 2011 Jun;11(6):389. doi: 10.1038/nrc3070. Epub 2011 May 12. Nat Rev Cancer. 2011. PMID: 21562582 No abstract available.
References
-
- Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–1018. - PubMed
-
- Adams PD. Healing and hurting: molecular mechanisms, functions, and pathologies of cellular senescence. Mol Cell. 2009;36:2–14. - PubMed
-
- Altomare DA, Tanno S, De Rienzo A, Klein-Szanto AJ, Skele KL, Hoffman JP, Testa JR. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J Cell Biochem. 2002;87:470–476. - PubMed
-
- Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004;23:8571–8580. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
