

Catalase and superoxide dismutase conjugated with platelet-endothelial cell adhesion molecule antibody distinctly alleviate abnormal endothelial permeability caused by exogenous reactive oxygen species and vascular endothelial growth factor
- PMID: 21474567
- PMCID: PMC3126647
- DOI: 10.1124/jpet.111.180620
Catalase and superoxide dismutase conjugated with platelet-endothelial cell adhesion molecule antibody distinctly alleviate abnormal endothelial permeability caused by exogenous reactive oxygen species and vascular endothelial growth factor
Abstract
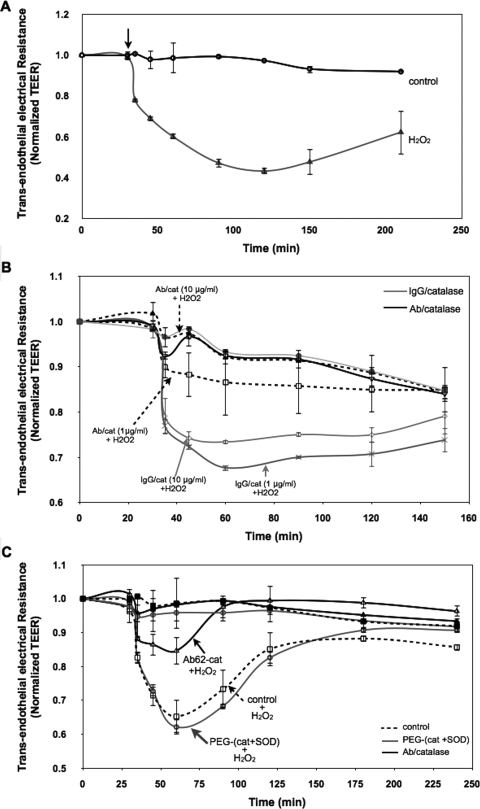
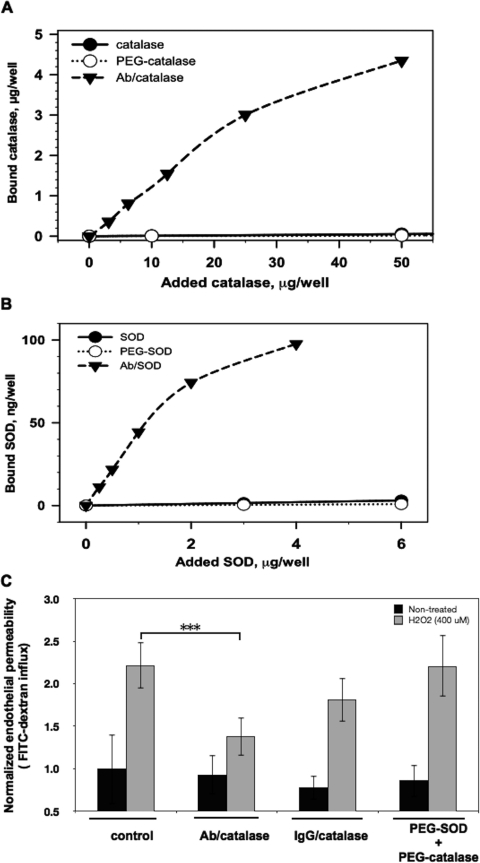
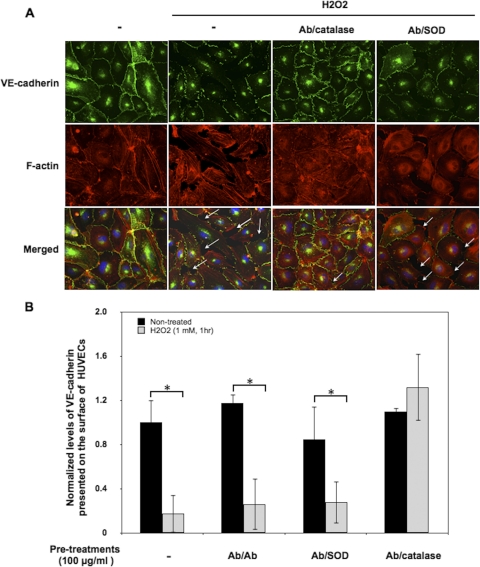
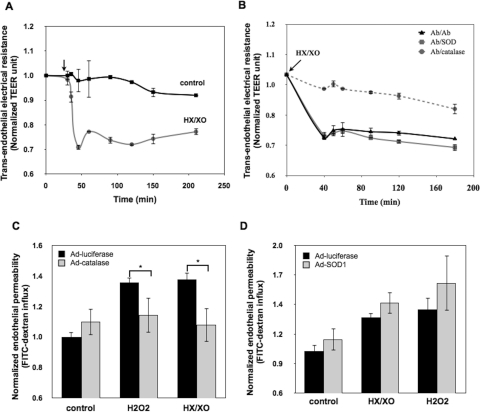
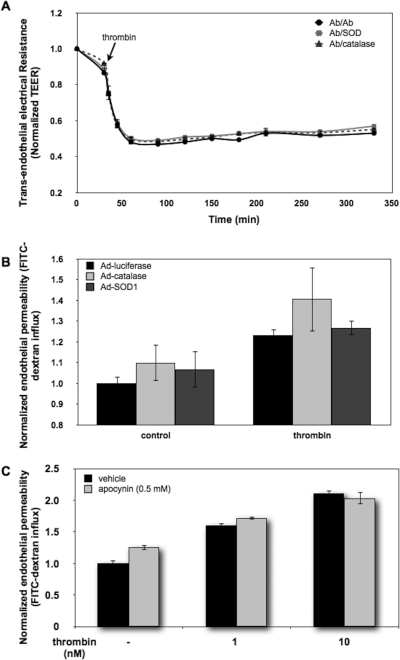
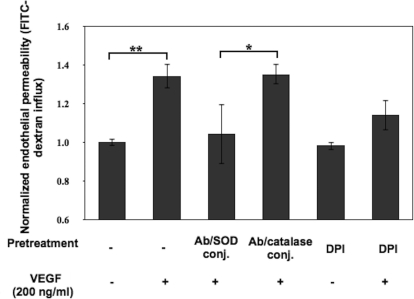
Reactive oxygen species (ROS) superoxide anion (O(2)()) and hydrogen peroxide (H(2)O(2)) produced by activated leukocytes and endothelial cells in sites of inflammation or ischemia cause endothelial barrier dysfunction that may lead to tissue edema. Antioxidant enzymes (AOEs) catalase and superoxide dismutase (SOD) conjugated with antibodies to platelet-endothelial cell adhesion molecule-1 (PECAM-1) specifically bind to endothelium, quench the corresponding ROS, and alleviate vascular oxidative stress and inflammation. In the present work, we studied the effects of anti-PECAM/catalase and anti-PECAM/SOD conjugates on the abnormal permeability manifested by transendothelial electrical resistance decline, increased fluorescein isothiocyanate-dextran influx, and redistribution of vascular endothelial-cadherin in human umbilical vein endothelial cell (HUVEC) monolayers. Anti-PECAM/catalase protected HUVEC monolayers against H(2)O(2)-induced endothelial barrier dysfunction. Polyethylene glycol-conjugated catalase exerted orders of magnitude lower endothelial uptake and no protective effect, similarly to IgG/catalase. Anti-PECAM/catalase, but not anti-PECAM/SOD, alleviated endothelial hyperpermeability caused by exposure to hypoxanthine/xanthine oxidase, implicating primarily H(2)O(2) in the disruption of the endothelial barrier in this model. Thrombin-induced endothelial permeability was not affected by treatment with anti-PECAM/AOEs or the NADPH oxidase inhibitor apocynin or overexpression of AOEs, indicating that the endogenous ROS play no key role in thrombin-mediated endothelial barrier dysfunction. In contrast, anti-PECAM/SOD, but not anti-PECAM/catalase, inhibited a vascular endothelial growth factor (VEGF)-induced increase in endothelial permeability, identifying a key role of endogenous O(2)() in the VEGF-mediated regulation of endothelial barrier function. Therefore, AOEs targeted to endothelial cells provide versatile molecular tools for testing the roles of specific ROS in vascular pathology and may be translated into remedies for these ROS-induced abnormalities.
Figures
References
-
- Alexander JS, Alexander BC, Eppihimer LA, Goodyear N, Haque R, Davis CP, Kalogeris TJ, Carden DL, Zhu YN, Kevil CG. (2000) Inflammatory mediators induce sequestration of VE-cadherin in cultured human endothelial cells. Inflammation 24:99–113 - PubMed
-
- Alom-Ruiz SP, Anilkumar N, Shah AM. (2008) Reactive oxygen species and endothelial activation. Antioxid Redox Signal 10:1089–1100 - PubMed
-
- Atochina EN, Balyasnikova IV, Danilov SM, Granger DN, Fisher AB, Muzykantov VR. (1998) Immunotargeting of catalase to ACE or ICAM-1 protects perfused rat lungs against oxidative stress. Am J Physiol 275:L806–L817 - PubMed
-
- Bae JS, Yang L, Manithody C, Rezaie AR. (2007) Engineering a disulfide bond to stabilize the calcium-binding loop of activated protein C eliminates its anticoagulant but not its protective signaling properties. J Biol Chem 282:9251–9259 - PubMed
-
- Barnard ML, Matalon S. (1992) Mechanisms of extracellular reactive oxygen species injury to the pulmonary microvasculature. J Appl Physiol 72:1724–1729 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
