

Rigorous treatment of multispecies multimode ligand-receptor interactions in 3D-QSAR: CoMFA analysis of thyroxine analogs binding to transthyretin
- PMID: 21476521
- PMCID: PMC3223556
- DOI: 10.1021/ci200055s
Rigorous treatment of multispecies multimode ligand-receptor interactions in 3D-QSAR: CoMFA analysis of thyroxine analogs binding to transthyretin
Abstract
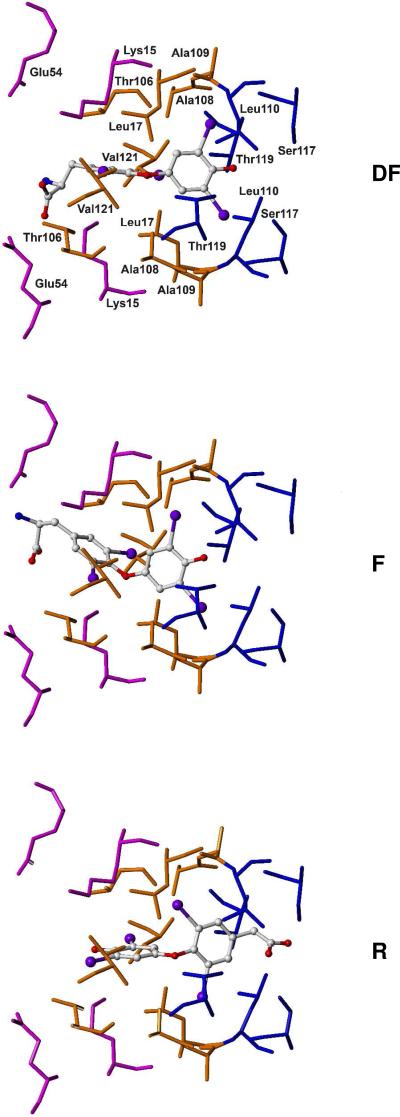
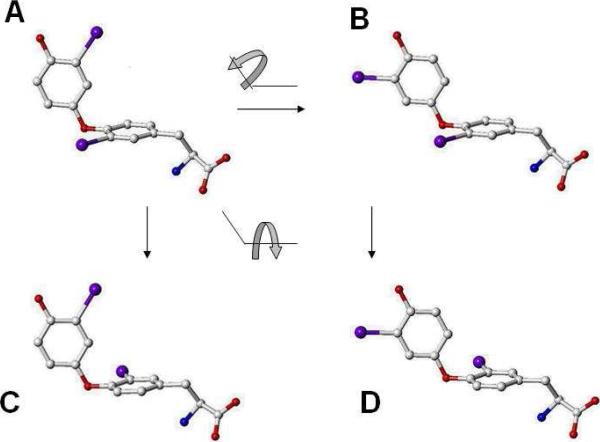
For a rigorous analysis of the receptor-ligand binding, speciation of the ligands caused by ionization, tautomerism, covalent hydration, and dynamic stereoisomerism needs to be considered. Each species may bind in several orientations or conformations (modes), especially for flexible ligands and receptors. A thermodynamic description of the multispecies (MS), multimode (MM) binding events shows that the overall association constant is equal to the weighted sum of the sums of microscopic association constants of individual modes for each species, with the weights given by the unbound fractions of individual species. This expression is a prerequisite for a precise quantitative characterization of the ligand-receptor interactions in both structure-based and ligand-based structure-activity analyses. We have implemented the MS-MM correlation expression into the comparative molecular field analysis (CoMFA), which deduces a map of the binding site from structures and binding affinities of a ligand set, in the absence of experimental structural information on the receptor. The MS-MM CoMFA approach was applied to published data for binding to transthyretin of 28 thyroxine analogs, each forming up to four ionization species under physiological conditions. The published X-ray structures of several analogs, exhibiting multiple binding modes, served as templates for the MS-MM superposition of thyroxine analogs. Additional modes were generated for compounds with flexible alkyl substituents, to identify bound conformations. The results demonstrate that the MS-MM modification improved predictive abilities of the CoMFA models, even for the standard procedure with MS-MM selected species and modes. The predicted prevalences of individual modes and the generated receptor site model are in reasonable agreement with the available X-ray data. The calibrated model can help in the design of inhibitors of transthyretin amyloid fibril formation.
Figures








References
-
- Hilal SH, Bornander LL, Carreira LA. Hydration equilibrium constants of aldehydes, ketones and quinazolines. QSAR & Combinatorial Science. 2005;24:631–637.
-
- Davies NM. Chiral inversion. Chirality Drug Des. Dev. 2004:351–392.
-
- Mattos C, Ringe D. Multiple binding modes. In: Kubinyi Hugo., editor. 3D QSAR in Drug Design: Theory, Methods, and Applications. Escom; Leiden: 1993. pp. 226–254.
-
- Case DA, Karplus M. Dynamics of ligand binding to heme protein. J. Mol. Biol. 1979;132:343–368. - PubMed
-
- Nelson RD, Gottlieb DI, Balasubramanian TM, Marshall GR. Opioid peptides: analysis of specificity and multiple binding modes through computer-aided drug design and structure-activity studies. NIDA Res. Monogr. 1986;69:204–230. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
