

Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1
- PMID: 21478864
- PMCID: PMC3087843
- DOI: 10.1038/nsmb.2021
Structural basis for antigenic peptide precursor processing by the endoplasmic reticulum aminopeptidase ERAP1
Abstract
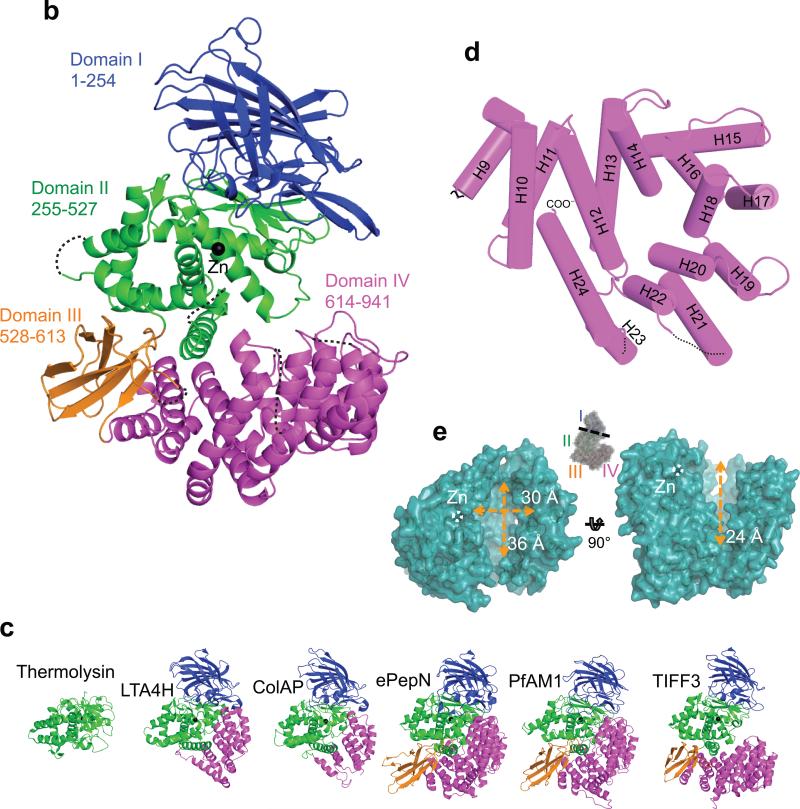
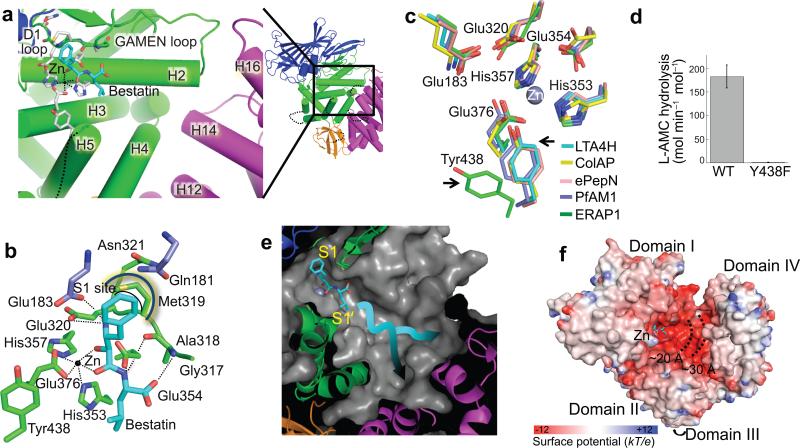
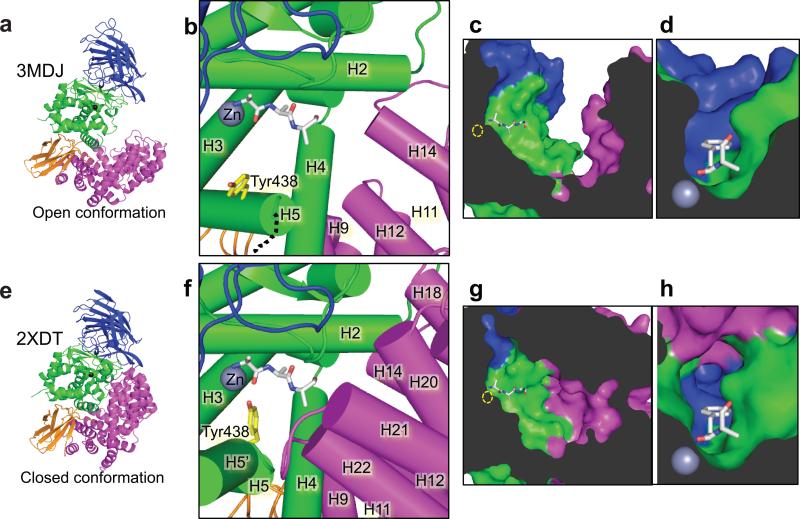
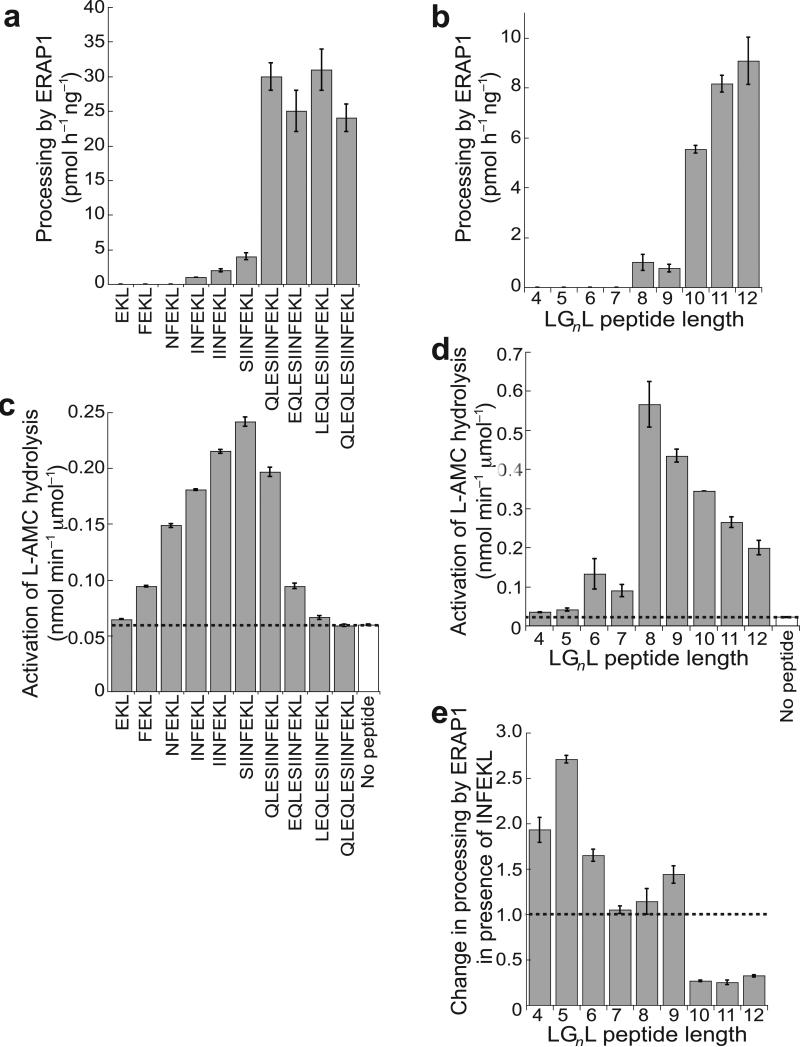
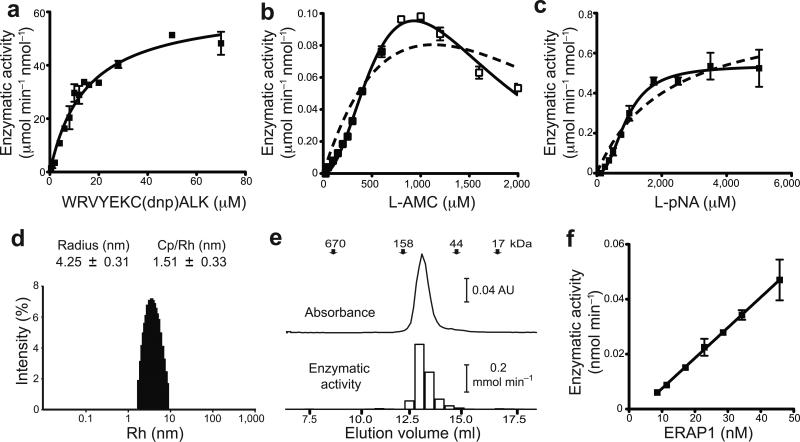
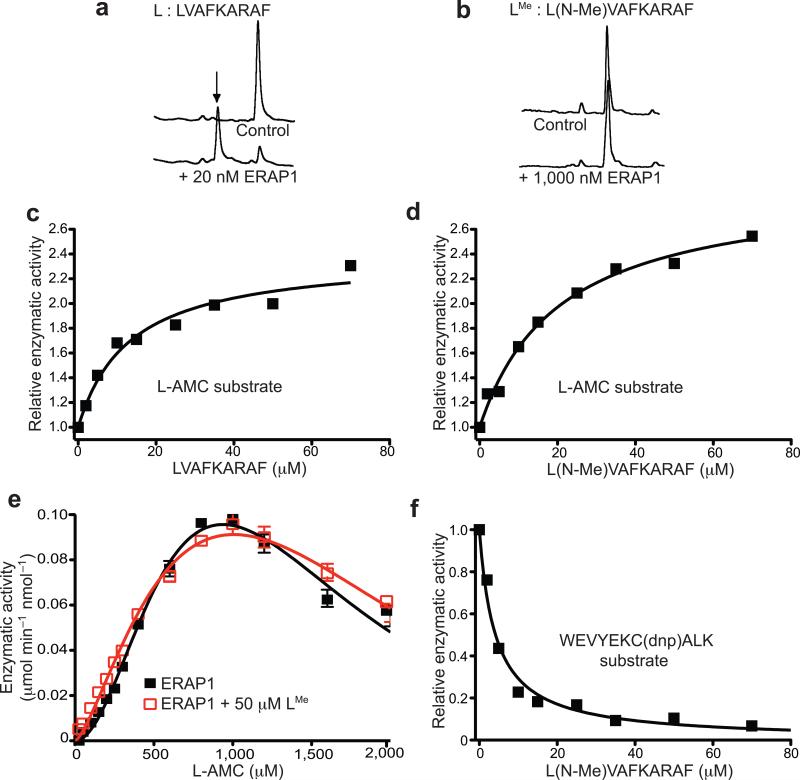
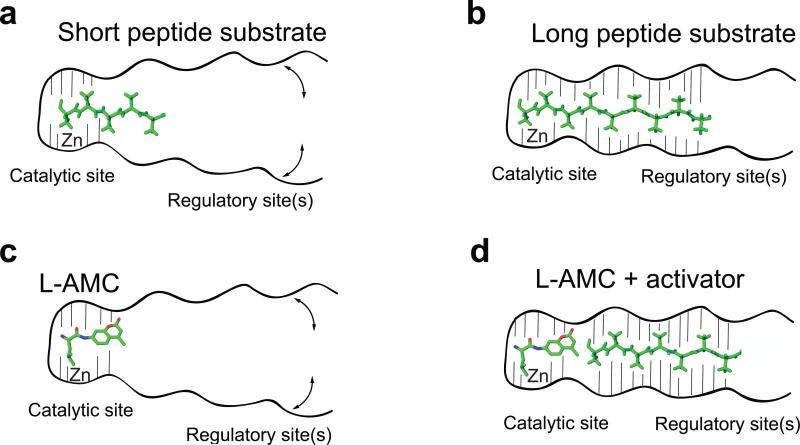
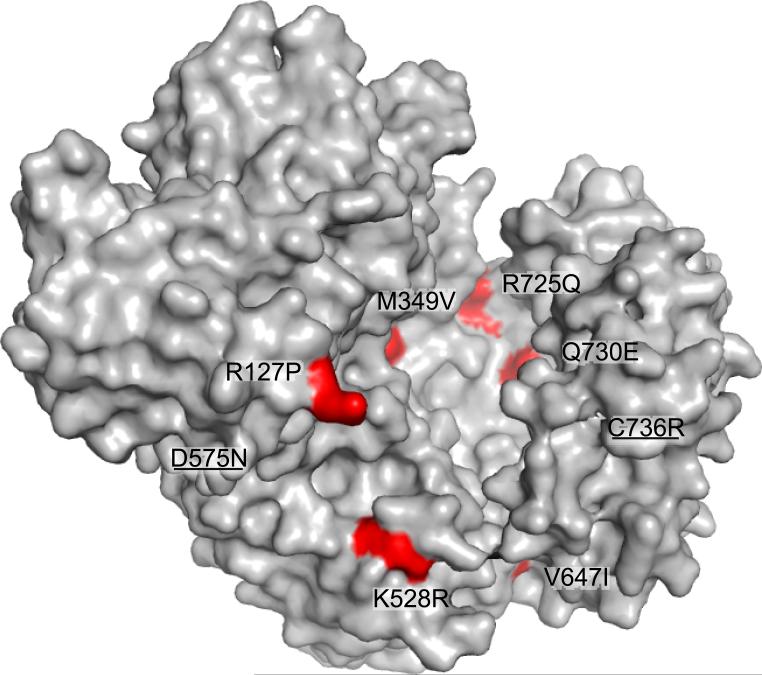
ERAP1 trims antigen precursors to fit into MHC class I proteins. To fulfill this function, ERAP1 has unique substrate preferences, trimming long peptides but sparing shorter ones. To identify the structural basis for ERAP1's unusual properties, we determined the X-ray crystal structure of human ERAP1 bound to bestatin. The structure reveals an open conformation with a large interior compartment. An extended groove originating from the enzyme's catalytic center can accommodate long peptides and has features that explain ERAP1's broad specificity for antigenic peptide precursors. Structural and biochemical analyses suggest a mechanism for ERAP1's length-dependent trimming activity, whereby binding of long rather than short substrates induces a conformational change with reorientation of a key catalytic residue toward the active site. ERAP1's unique structural elements suggest how a generic aminopeptidase structure has been adapted for the specialized function of trimming antigenic precursors.
Figures

References
-
- Rock KL, York IA, Goldberg AL. Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat Immunol. 2004;5:670–7. - PubMed
-
- Fruci D, Niedermann G, Butler RH, van Endert PM. Efficient MHC class I-independent amino-terminal trimming of epitope precursor peptides in the endoplasmic reticulum. Immunity. 2001;15:467–76. - PubMed
-
- Serwold T, Gaw S, Shastri N. ER aminopeptidases generate a unique pool of peptides for MHC class I molecules. Nat Immunol. 2001;2:644–51. - PubMed
-
- Saric T, et al. An IFN-gamma-induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I-presented peptides. Nat Immunol. 2002;3:1169–76. - PubMed
-
- Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. ERAAP customizes peptides for MHC class I molecules in the endoplasmic reticulum. Nature. 2002;419:480–3. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
