

The Arabidopsis lyrata genome sequence and the basis of rapid genome size change
- PMID: 21478890
- PMCID: PMC3083492
- DOI: 10.1038/ng.807
The Arabidopsis lyrata genome sequence and the basis of rapid genome size change
Abstract
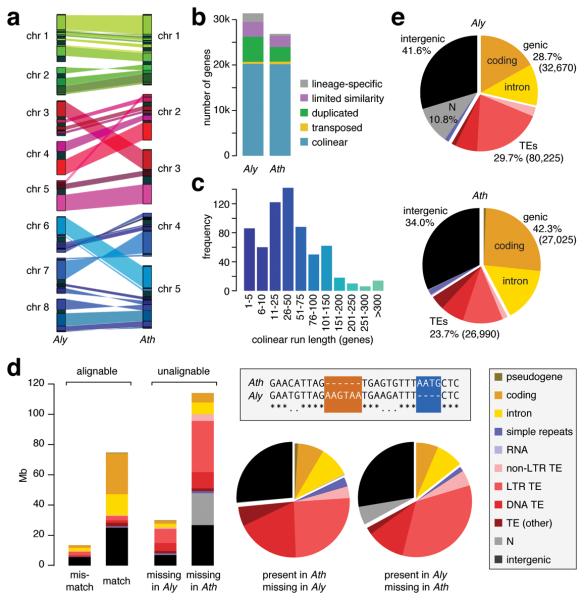
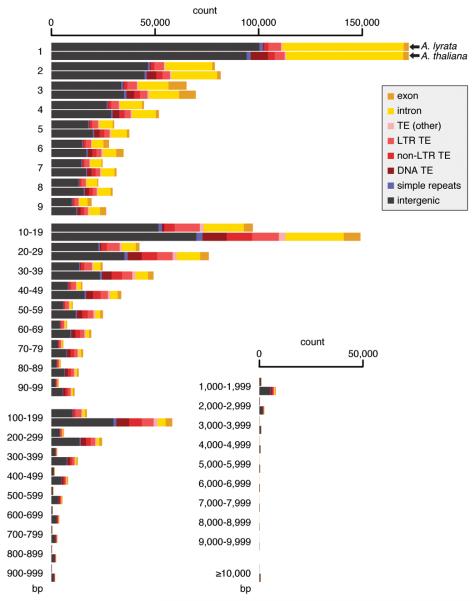
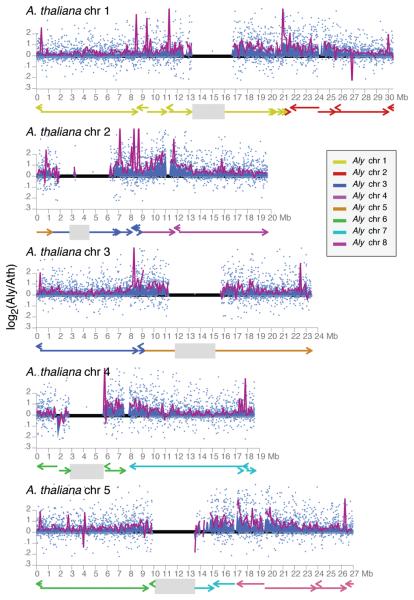
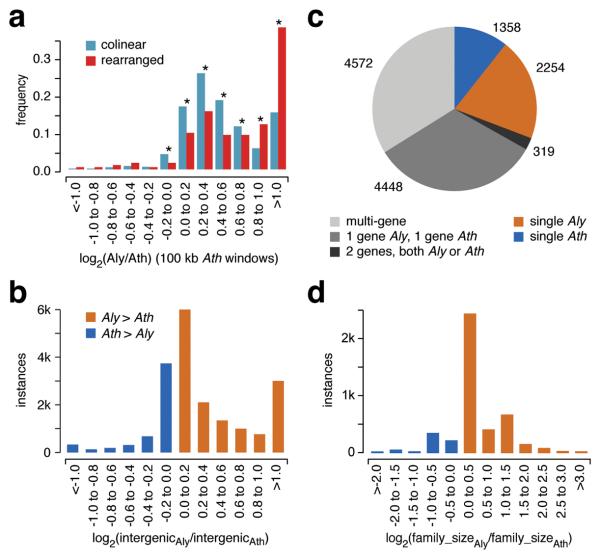
We report the 207-Mb genome sequence of the North American Arabidopsis lyrata strain MN47 based on 8.3× dideoxy sequence coverage. We predict 32,670 genes in this outcrossing species compared to the 27,025 genes in the selfing species Arabidopsis thaliana. The much smaller 125-Mb genome of A. thaliana, which diverged from A. lyrata 10 million years ago, likely constitutes the derived state for the family. We found evidence for DNA loss from large-scale rearrangements, but most of the difference in genome size can be attributed to hundreds of thousands of small deletions, mostly in noncoding DNA and transposons. Analysis of deletions and insertions still segregating in A. thaliana indicates that the process of DNA loss is ongoing, suggesting pervasive selection for a smaller genome. The high-quality reference genome sequence for A. lyrata will be an important resource for functional, evolutionary and ecological studies in the genus Arabidopsis.
Figures
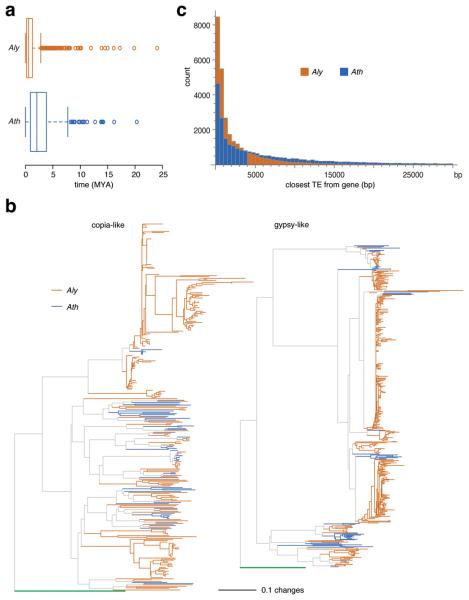
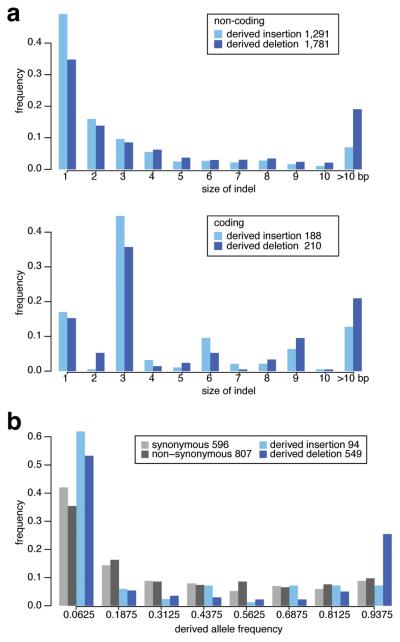
References
-
- Greilhuber J, et al. Smallest angiosperm genomes found in Lentibulariaceae, with chromosomes of bacterial size. Plant Biol. 2006;8:770–7. - PubMed
-
- Gaut BS, Ross-Ibarra J. Selection on major components of angiosperm genomes. Science. 2008;320:484–6. - PubMed
-
- Pellicer J, Fay MF, Leitch IJ. The largest eukaryotic genome of them all? Botanical Journal of the Linnean Society. 2010;164:10–15.
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
