

Integrated analysis of multiple microarray datasets identifies a reproducible survival predictor in ovarian cancer
- PMID: 21479231
- PMCID: PMC3066217
- DOI: 10.1371/journal.pone.0018202
Integrated analysis of multiple microarray datasets identifies a reproducible survival predictor in ovarian cancer
Abstract
Background: Public data integration may help overcome challenges in clinical implementation of microarray profiles. We integrated several ovarian cancer datasets to identify a reproducible predictor of survival.
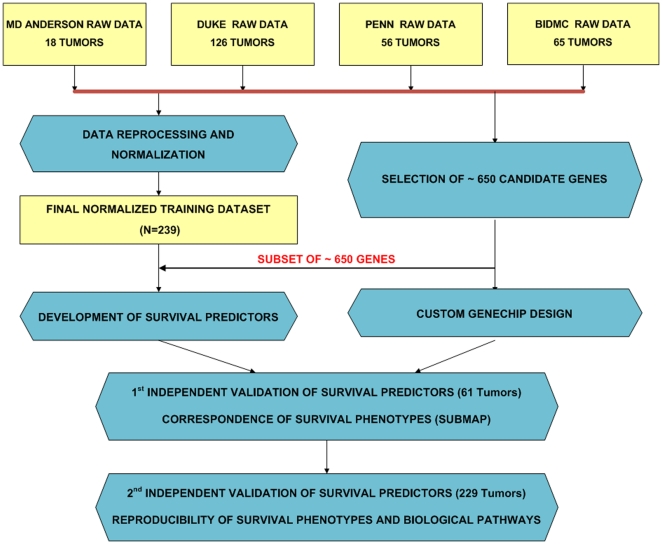
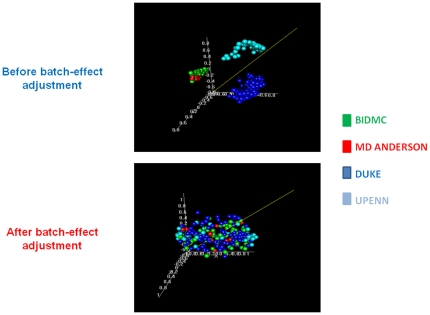
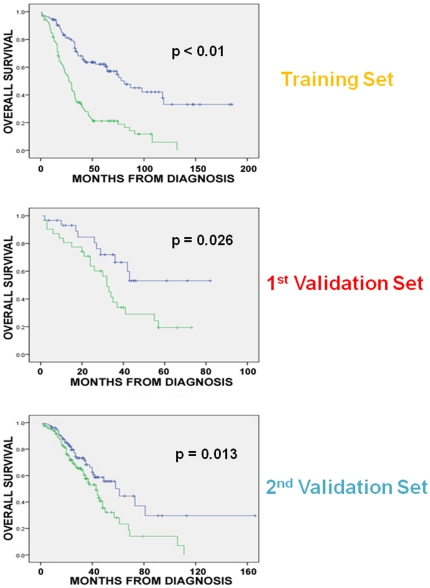
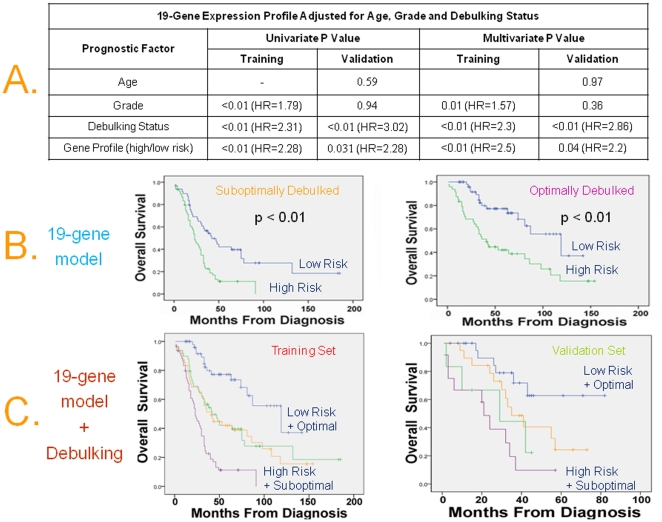
Methodology/principal findings: Four microarray datasets from different institutions comprising 265 advanced stage tumors were uniformly reprocessed into a single training dataset, also adjusting for inter-laboratory variation ("batch-effect"). Supervised principal component survival analysis was employed to identify prognostic models. Models were independently validated in a 61-patient cohort using a custom array genechip and a publicly available 229-array dataset. Molecular correspondence of high- and low-risk outcome groups between training and validation datasets was demonstrated using Subclass Mapping. Previously established molecular phenotypes in the 2(nd) validation set were correlated with high and low-risk outcome groups. Functional representational and pathway analysis was used to explore gene networks associated with high and low risk phenotypes. A 19-gene model showed optimal performance in the training set (median OS 31 and 78 months, p < 0.01), 1(st) validation set (median OS 32 months versus not-yet-reached, p = 0.026) and 2(nd) validation set (median OS 43 versus 61 months, p = 0.013) maintaining independent prognostic power in multivariate analysis. There was strong molecular correspondence of the respective high- and low-risk tumors between training and 1(st) validation set. Low and high-risk tumors were enriched for favorable and unfavorable molecular subtypes and pathways, previously defined in the public 2(nd) validation set.
Conclusions/significance: Integration of previously generated cancer microarray datasets may lead to robust and widely applicable survival predictors. These predictors are not simply a compilation of prognostic genes but appear to track true molecular phenotypes of good- and poor-outcome.
Conflict of interest statement
Figures

References
-
- Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519–2529. - PubMed
-
- McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334:1–6. - PubMed
-
- Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21:3194–3200. - PubMed
-
- Bristow RE, Tomacruz RS, Armstrong DK, Trimble EL, Montz FJ. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: a meta-analysis. J Clin Oncol. 2002;20:1248–1259. - PubMed
-
- Thigpen T, Brady MF, Omura GA, Creasman WT, McGuire WP, et al. Age as a prognostic factor in ovarian carcinoma. The Gynecologic Oncology Group experience. Cancer. 1993;71:606–614. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
