

A missense mutation in Fgfr1 causes ear and skull defects in hush puppy mice
- PMID: 21479780
- PMCID: PMC3099004
- DOI: 10.1007/s00335-011-9324-8
A missense mutation in Fgfr1 causes ear and skull defects in hush puppy mice
Abstract
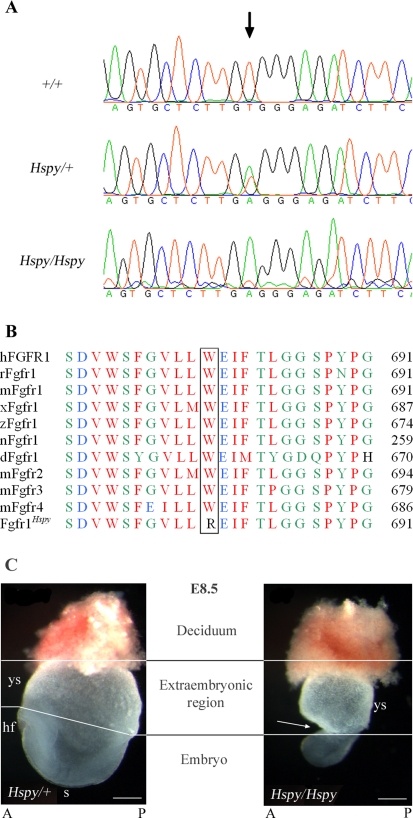
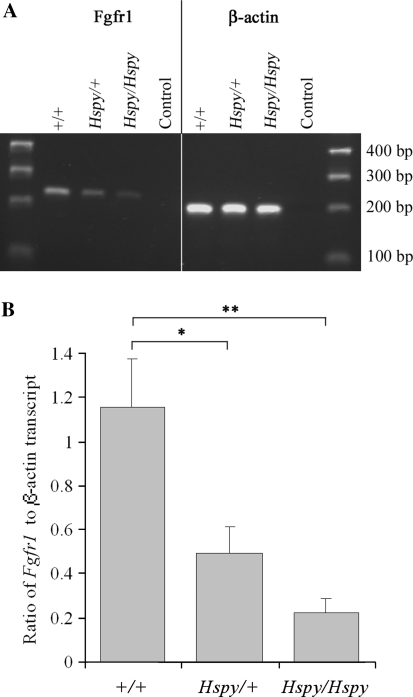
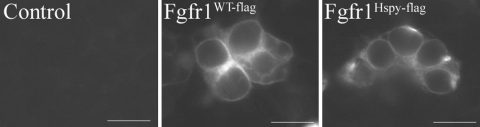
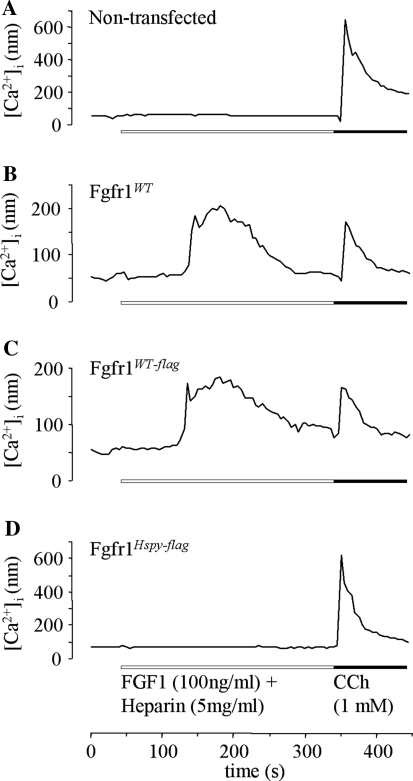
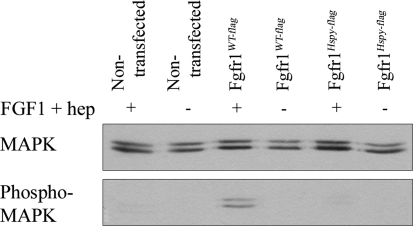
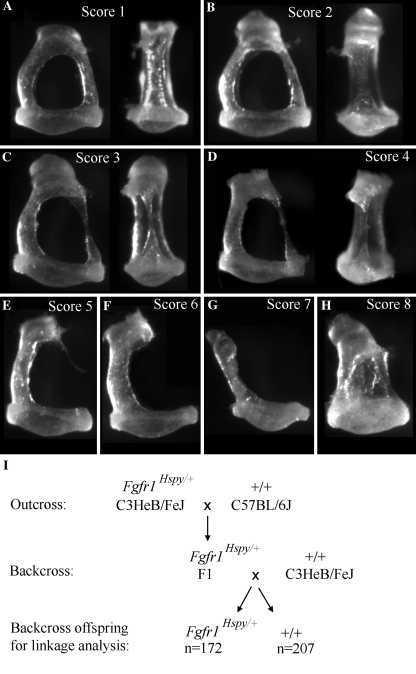
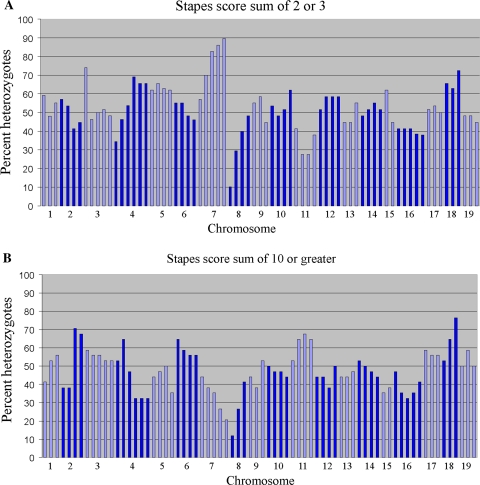
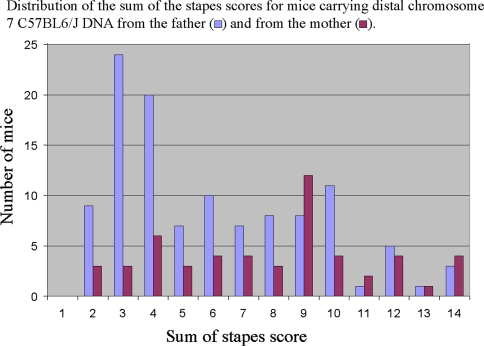
The hush puppy mouse mutant has been shown previously to have skull and outer, middle, and inner ear defects, and an increase in hearing threshold. The fibroblast growth factor receptor 1 (Fgfr1) gene is located in the region of chromosome 8 containing the mutation. Sequencing of the gene in hush puppy heterozygotes revealed a missense mutation in the kinase domain of the protein (W691R). Homozygotes were found to die during development, at approximately embryonic day 8.5, and displayed a phenotype similar to null mutants. Reverse transcription PCR indicated a decrease in Fgfr1 transcript in heterozygotes and homozygotes. Generation of a construct containing the mutation allowed the function of the mutated receptor to be studied. Immunocytochemistry showed that the mutant receptor protein was present at the cell membrane, suggesting normal expression and trafficking. Measurements of changes in intracellular calcium concentration showed that the mutated receptor could not activate the IP(3) pathway, in contrast to the wild-type receptor, nor could it initiate activation of the Ras/MAP kinase pathway. Thus, the hush puppy mutation in fibroblast growth factor receptor 1 appears to cause a loss of receptor function. The mutant protein appears to have a dominant negative effect, which could be due to it dimerising with the wild-type protein and inhibiting its activity, thus further reducing the levels of functional protein. A dominant modifier, Mhspy, which reduces the effect of the hush puppy mutation on pinna and stapes development, has been mapped to the distal end of chromosome 7 and may show imprinting.
Figures
Similar articles
-
Hush puppy: a new mouse mutant with pinna, ossicle, and inner ear defects.Laryngoscope. 2005 Jan;115(1):116-24. doi: 10.1097/01.mlg.0000150693.31130.a0. Laryngoscope. 2005. PMID: 15630379
-
A Pro250Arg substitution in mouse Fgfr1 causes increased expression of Cbfa1 and premature fusion of calvarial sutures.Hum Mol Genet. 2000 Aug 12;9(13):2001-8. doi: 10.1093/hmg/9.13.2001. Hum Mol Genet. 2000. PMID: 10942429
-
Missense mutation in the second RNA binding domain reveals a role for Prkra (PACT/RAX) during skull development.PLoS One. 2011;6(12):e28537. doi: 10.1371/journal.pone.0028537. Epub 2011 Dec 14. PLoS One. 2011. PMID: 22194846 Free PMC article.
-
Regulation of fibroblast growth factor receptor-1 (FGFR1) by thyroid hormone: identification of a thyroid hormone response element in the murine Fgfr1 promoter.Endocrinology. 2007 Dec;148(12):5966-76. doi: 10.1210/en.2007-0114. Epub 2007 Aug 30. Endocrinology. 2007. PMID: 17761769
-
Clinical findings in a patient with FGFR1 P252R mutation and comparison with the literature.Am J Med Genet. 2000 Jul 3;93(1):22-8. doi: 10.1002/1096-8628(20000703)93:1<22::aid-ajmg5>3.0.co;2-u. Am J Med Genet. 2000. PMID: 10861678 Review.
Cited by
-
Exome sequencing identifies a missense mutation in Isl1 associated with low penetrance otitis media in dearisch mice.Genome Biol. 2011 Sep 21;12(9):R90. doi: 10.1186/gb-2011-12-9-r90. Genome Biol. 2011. PMID: 21936904 Free PMC article.
-
An Essential Requirement for Fgf10 in Pinna Extension Sheds Light on Auricle Defects in LADD Syndrome.Front Cell Dev Biol. 2020 Dec 10;8:609643. doi: 10.3389/fcell.2020.609643. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 33363172 Free PMC article.
-
Mature middle and inner ears express Chd7 and exhibit distinctive pathologies in a mouse model of CHARGE syndrome.Hear Res. 2011 Dec;282(1-2):184-95. doi: 10.1016/j.heares.2011.08.005. Epub 2011 Aug 23. Hear Res. 2011. PMID: 21875659 Free PMC article.
-
FGF/FGFR signaling in health and disease.Signal Transduct Target Ther. 2020 Sep 2;5(1):181. doi: 10.1038/s41392-020-00222-7. Signal Transduct Target Ther. 2020. PMID: 32879300 Free PMC article. Review.
-
Uncovering the secreted signals and transcription factors regulating the development of mammalian middle ear ossicles.Dev Dyn. 2020 Dec;249(12):1410-1424. doi: 10.1002/dvdy.260. Epub 2020 Oct 28. Dev Dyn. 2020. PMID: 33058336 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
