

A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in MYH14
- PMID: 21480433
- PMCID: PMC3103632
- DOI: 10.1002/humu.21488
A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in MYH14
Abstract
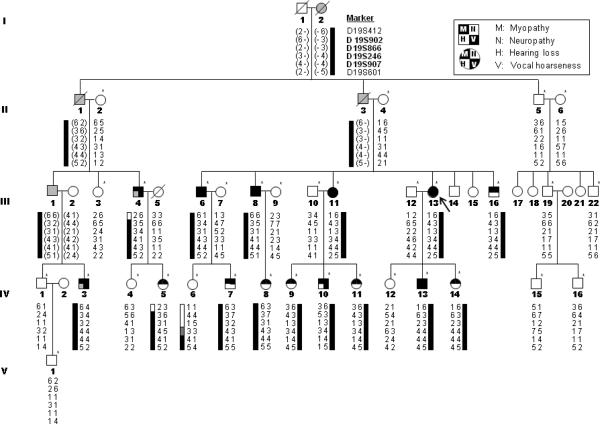
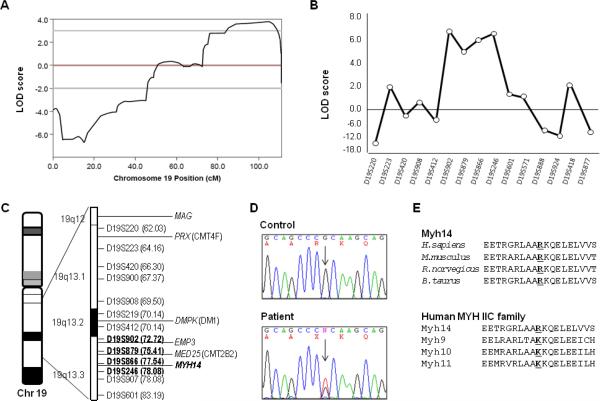
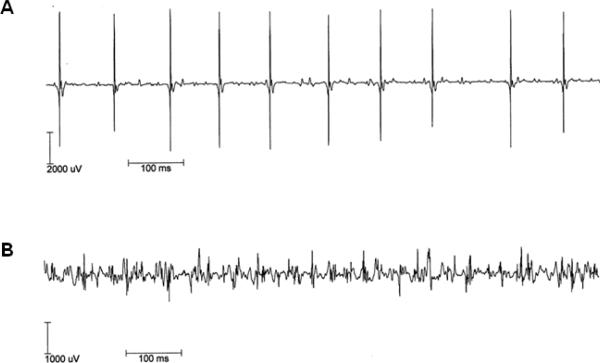
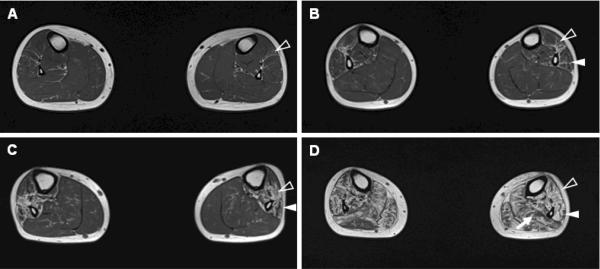
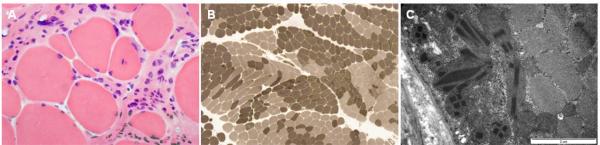
Both peripheral neuropathy and distal myopathy are well-established inherited neuromuscular disorders characterized by progressive weakness and atrophy of the distal limb muscles. A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss was diagnosed in a large autosomal dominant Korean family. A high density single nucleotide polymorphism (SNP)-based linkage study mapped the underlying gene to a region on chromosome 19q13.3. The maximum multipoint LOD score was 3.794. Sequencing of 34 positional candidate genes in the segregating haplotype revealed a novel c.2822G>T (p.Arg941Leu) mutation in the gene MYH14, which encodes the nonmuscle myosin heavy chain 14. Clinically we observed a sequential pattern of the onset of muscle weakness starting from the anterior to the posterior leg muscle compartments followed by involvement of intrinsic hand and proximal muscles. The hearing loss and hoarseness followed the onset of distal muscle weakness. Histopathologic and electrodiagnostic studies revealed both chronic neuropathic and myopathic features in the affected patients. Although mutations in MYH14 have been shown to cause nonsyndromic autosomal dominant hearing loss (DFNA4), the peripheral neuropathy, myopathy, and hoarseness have not been associated with MYH14. Therefore, we suggest that the identified mutation in MYH14 significantly expands the phenotypic spectrum of this gene.
© 2011 Wiley-Liss, Inc.
Figures
References
-
- Arias M, Pardo J, Blanco-Arias P, Sobrido MJ, Arias S, Dapena D, Carracedo A, Goldfarb LG, Navarro C. Distinct phenotypic features and gender-specific disease manifestations in a Spanish family with desmin L370P mutation. Neuromuscul Disord. 2006;16:498–503. - PubMed
-
- Astuto LM, Kelley PM, Askew JW, Weston MD, Smith RJ, Alswaid AF, Al-Rakaf M, Kimberling WJ. Searching for evidence of DFNB2. Am J Med Genet. 2002;109:291–297. - PubMed
-
- Berghoff C, Berghoff M, Leal A, Morera B, Barrantes R, Reis A, Neundörfer B, Rautenstrauss B, Del Valle G, Heuss D. Clinical and electrophysiological characteristics of autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2B) that maps to chromosome 19q13.3. Neuromuscul Disord. 2004;5:301–306. - PubMed
-
- Bergmann C, Senderek J, Hermanns B, Jauch A, Janssen B, Schröder JM, Karch D. Becker muscular dystrophy combined with X-linked Charcot-Marie-Tooth neuropathy. Muscle Nerve. 2000;23:818–823. - PubMed
-
- Braida C, Stefanatos RK, Adam B, Mahajan N, Smeets HJ, Niel F, Goizet C, Arveiler B, Koenig M, Lagier-Tourenne C, et al. Variant CCG and GGC repeats within the CTG expansion dramatically modify mutational dynamics and likely contribute toward unusual symptoms in some myotonic dystrophy type 1 patients. Hum Mol Genet. 2010;19:1399–1412. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
