

Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed
- PMID: 21480434
- PMCID: PMC4154965
- DOI: 10.1002/humu.21490
Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed
Abstract
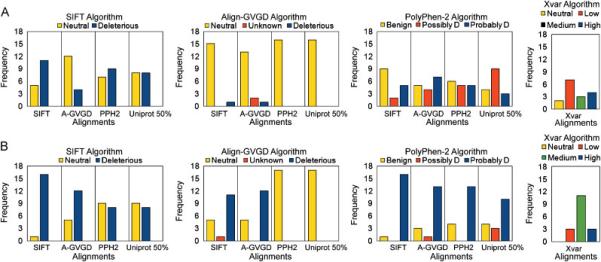
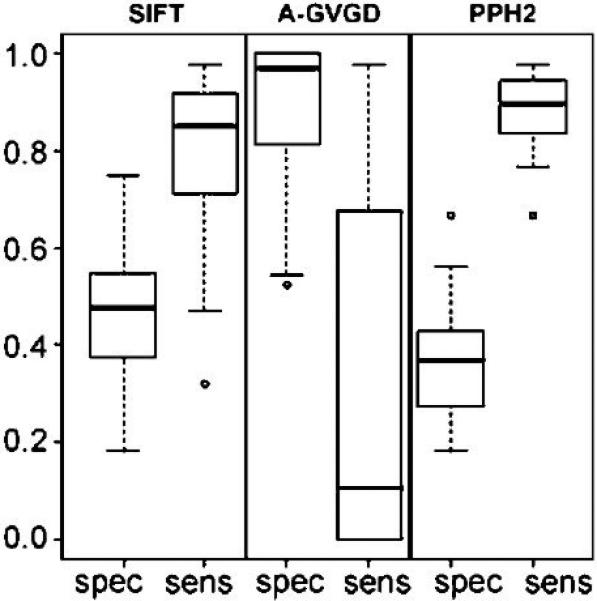
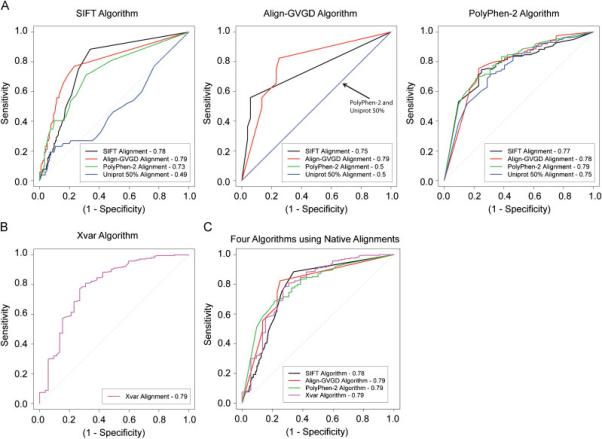
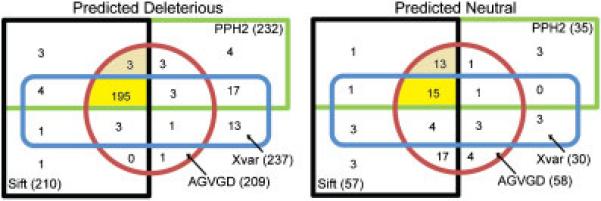
Multiple algorithms are used to predict the impact of missense mutations on protein structure and function using algorithm-generated sequence alignments or manually curated alignments. We compared the accuracy with native alignment of SIFT, Align-GVGD, PolyPhen-2, and Xvar when generating functionality predictions of well-characterized missense mutations (n = 267) within the BRCA1, MSH2, MLH1, and TP53 genes. We also evaluated the impact of the alignment employed on predictions from these algorithms (except Xvar) when supplied the same four alignments including alignments automatically generated by (1) SIFT, (2) Polyphen-2, (3) Uniprot, and (4) a manually curated alignment tuned for Align-GVGD. Alignments differ in sequence composition and evolutionary depth. Data-based receiver operating characteristic curves employing the native alignment for each algorithm result in area under the curve of 78-79% for all four algorithms. Predictions from the PolyPhen-2 algorithm were least dependent on the alignment employed. In contrast, Align-GVGD predicts all variants neutral when provided alignments with a large number of sequences. Of note, algorithms make different predictions of variants even when provided the same alignment and do not necessarily perform best using their own alignment. Thus, researchers should consider optimizing both the algorithm and sequence alignment employed in missense prediction.
© 2011 Wiley-Liss, Inc.
Figures
Comment in
-
Comparison of programs for in silico assessment of missense substitutions.Hum Mutat. 2011 Jun;32(6):v. doi: 10.1002/humu.21532. Hum Mutat. 2011. PMID: 21618348 No abstract available.
References
-
- Abramowitz M, Stegun IA. Handbook of Mathematical Functions with Formulas, Graphs, and Mathematical Tables. U.S. Government Printing Office; Washington, D.C.: 1972. p. 885.
-
- Agresti A. Categorical Data Analysis. 2nd edition. John Wiley and Sons; Hoboken, New Jersey: 2002.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
