

Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036
- PMID: 21481795
- PMCID: PMC3077923
- DOI: 10.1016/j.ccr.2011.03.003
Conformational control inhibition of the BCR-ABL1 tyrosine kinase, including the gatekeeper T315I mutant, by the switch-control inhibitor DCC-2036
Abstract
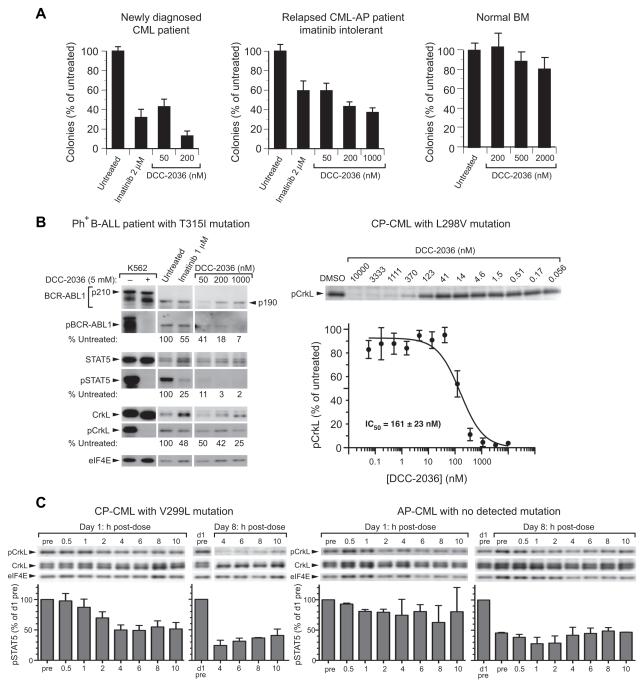
Acquired resistance to ABL1 tyrosine kinase inhibitors (TKIs) through ABL1 kinase domain mutations, particularly the gatekeeper mutant T315I, is a significant problem for patients with chronic myeloid leukemia (CML). Using structure-based drug design, we developed compounds that bind to residues (Arg386/Glu282) ABL1 uses to switch between inactive and active conformations. The lead "switch-control" inhibitor, DCC-2036, potently inhibits both unphosphorylated and phosphorylated ABL1 by inducing a type II inactive conformation, and retains efficacy against the majority of clinically relevant CML-resistance mutants, including T315I. DCC-2036 inhibits BCR-ABL1(T315I)-expressing cell lines, prolongs survival in mouse models of T315I mutant CML and B-lymphoblastic leukemia, and inhibits primary patient leukemia cells expressing T315I in vitro and in vivo, supporting its clinical development in TKI-resistant Ph(+) leukemia.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
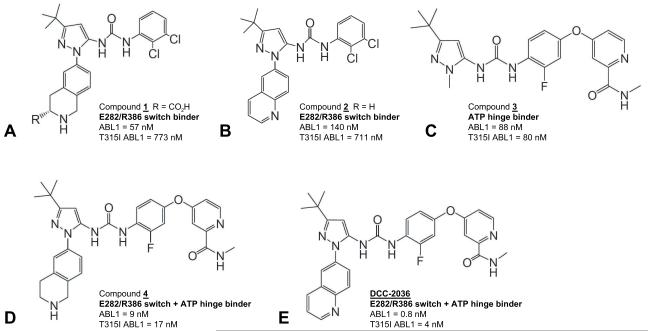
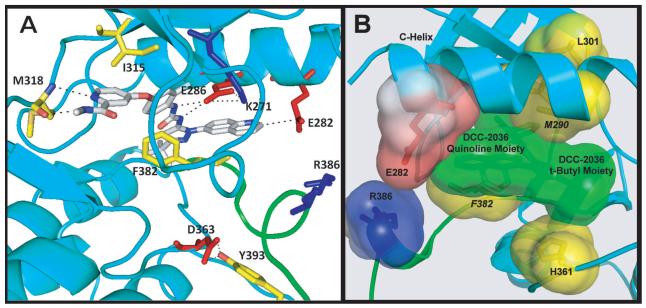
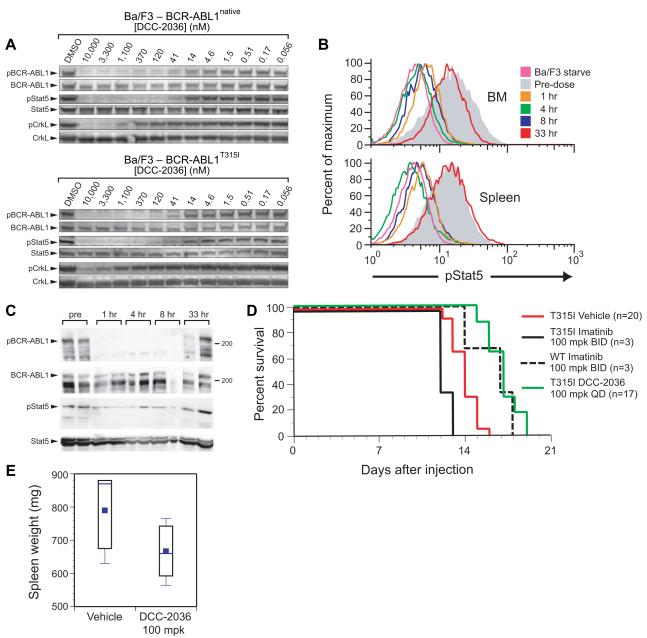
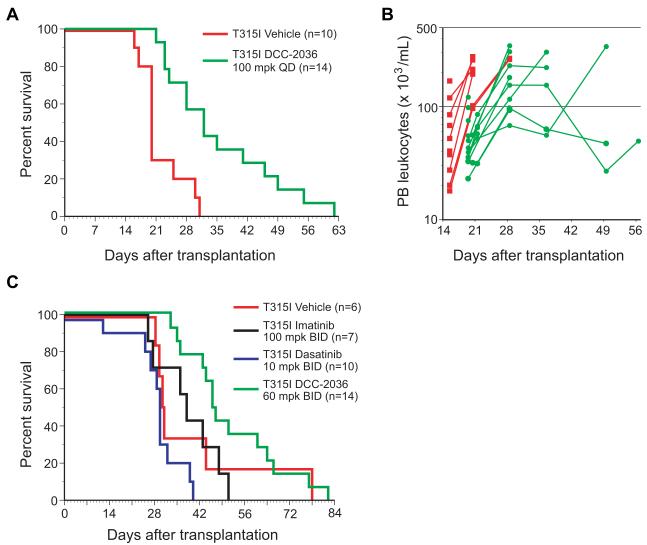
Comment in
-
Unswitch-ABL drugs overcome resistance in chronic myeloid leukemia.Cancer Cell. 2011 Apr 12;19(4):435-7. doi: 10.1016/j.ccr.2011.03.019. Cancer Cell. 2011. PMID: 21481785
-
Leukaemia: Now get out of that..Nat Rev Cancer. 2011 Jun;11(6):388. doi: 10.1038/nrc3067. Epub 2011 May 12. Nat Rev Cancer. 2011. PMID: 21562581 No abstract available.
-
Anticancer drugs: Now get out of that...Nat Rev Drug Discov. 2011 Jun;10(6):414-5. doi: 10.1038/nrd3471. Nat Rev Drug Discov. 2011. PMID: 21629288 No abstract available.
References
-
- Azam M, Latek RR, Daley GQ. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 2003;112:831–843. - PubMed
-
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. - PubMed
-
- Deininger MW, Druker BJ. Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol. Rev. 2003;55:401–423. - PubMed
-
- Donato NJ, Fang D, Sun H, Giannola D, Peterson LF, Talpaz M. Targets and effectors of the cellular response to aurora kinase inhibitor MK-0457 (VX-680) in imatinib sensitive and resistant chronic myelogenous leukemia. Biochem. Pharmacol. 2010;79:688–697. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
