

Endothelial cell activation by antiphospholipid antibodies is modulated by Kruppel-like transcription factors
- PMID: 21482710
- PMCID: PMC3122956
- DOI: 10.1182/blood-2010-10-313072
Endothelial cell activation by antiphospholipid antibodies is modulated by Kruppel-like transcription factors
Abstract
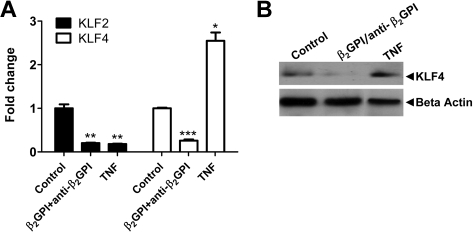
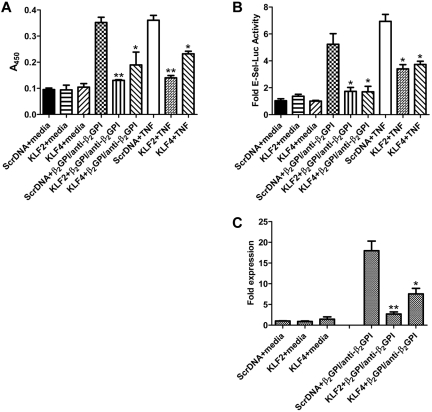
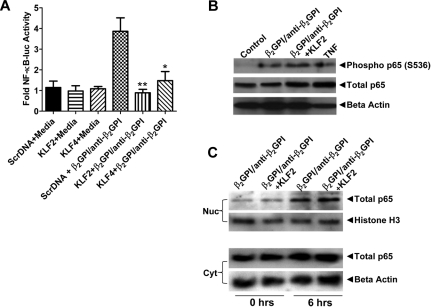
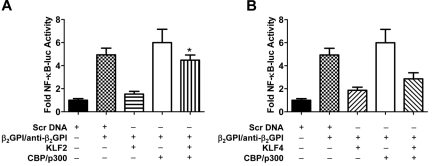
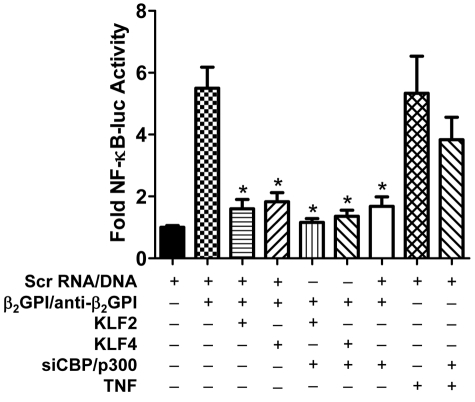
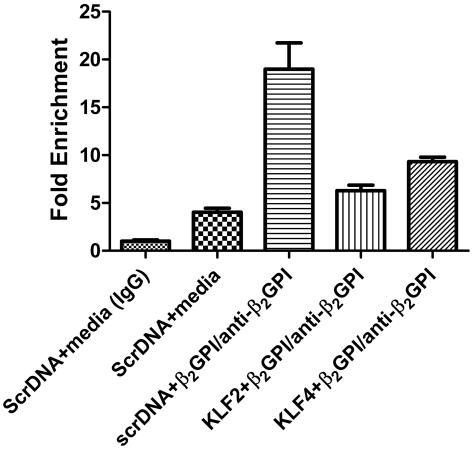
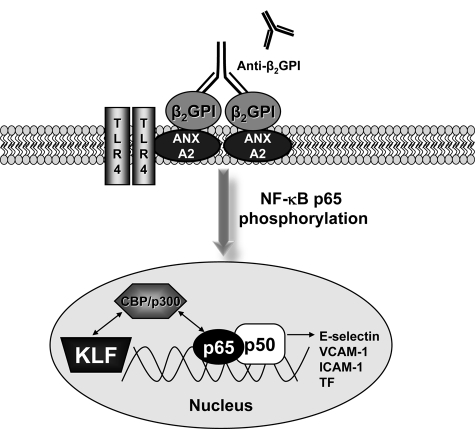
Antiphospholipid syndrome is characterized by thrombosis and/or recurrent pregnancy loss in the presence of antiphospholipid antibodies (APLAs). The majority of APLAs are directed against phospholipid-binding proteins, particularly β₂-glycoprotein I (β₂GPI). Anti-β₂GPI antibodies activate endothelial cells in a β₂GPI-dependent manner through a pathway that involves NF-κB. Krüppel-like factors (KLFs) play a critical role in regulating the endothelial response to inflammatory stimuli. We hypothesized that activation of endothelial cells by APLA/anti-β₂GPI antibodies might be associated with decreased expression of KLFs, which in turn might facilitate cellular activation mediated through NF-κB. Our experimental results confirmed this hypothesis, demonstrating markedly decreased expression of KLF2 and KLF4 after incubation of cells with APLA/anti-β₂GPI antibodies. Restoration of KLF2 or KLF4 levels inhibited NF-κB transcriptional activity and blocked APLA/anti-β₂GPI-mediated endothelial activation despite NF-κB p65 phosphorylation. Chromatin immunoprecipitation analysis demonstrated that inhibition of NF-κB transcriptional activity by KLFs reflects sequestration of the cotranscriptional activator CBP/p300, making this cofactor unavailable to NF-κB. These findings suggest that the endothelial response to APLA/anti-β₂GPI antibodies reflects competition between KLFs and NF-κB for their common cofactor, CBP/p300. Taken together, these observations are the first to implicate the KLFs as novel participants in the endothelial proinflammatory response to APLA/anti-β₂GPI antibodies.
Figures
References
-
- Arnout J, Vermylen J. Current status and implications of autoimmune antiphospholipid antibodies in relation to thrombotic disease. J Thromb Haemost. 2003;1(5):931–942. - PubMed
-
- Galli M, Barbui T. Antiphospholipid antibodies and thrombosis: strength of association. Hematol J. 2003;4(3):180–186. - PubMed
-
- Rand JH. The antiphospholipid syndrome. Annu Rev Med. 2003;54:409–424. - PubMed
-
- Giannakopoulos B, Passam F, Rahgozar S, Krilis SA. Current concepts on the pathogenesis of the antiphospholipid syndrome. Blood. 2007;109(2):422–430. - PubMed
-
- de Laat B, Eckmann CM, van Schagen M, Meijer AB, Mertens K, van Mourik JA. Correlation between the potency of a beta2-glycoprotein I-dependent lupus anticoagulant and the level of resistance to activated protein C. Blood Coagul Fibrinolysis. 2008;19(8):757–764. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K08HL083090/HL/NHLBI NIH HHS/United States
- R01 HL086548/HL/NHLBI NIH HHS/United States
- R01 HL097593/HL/NHLBI NIH HHS/United States
- R01 HL110630/HL/NHLBI NIH HHS/United States
- HL086548/HL/NHLBI NIH HHS/United States
- R01 HL084154/HL/NHLBI NIH HHS/United States
- K08 HL083090/HL/NHLBI NIH HHS/United States
- P50 HL081011/HL/NHLBI NIH HHS/United States
- HL084154/HL/NHLBI NIH HHS/United States
- T32HL007147/HL/NHLBI NIH HHS/United States
- R01 HL076754/HL/NHLBI NIH HHS/United States
- R01 HL119195/HL/NHLBI NIH HHS/United States
- R01 HL075427/HL/NHLBI NIH HHS/United States
- HL075427/HL/NHLBI NIH HHS/United States
- HL097593/HL/NHLBI NIH HHS/United States
- HL076754/HL/NHLBI NIH HHS/United States
- P50HL081011/HL/NHLBI NIH HHS/United States
- T32 HL007147/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
