

Amyloidosis: pathogenesis and new therapeutic options
- PMID: 21483018
- PMCID: PMC3138545
- DOI: 10.1200/JCO.2010.32.2271
Amyloidosis: pathogenesis and new therapeutic options
Abstract
The systemic amyloidoses are a group of complex diseases caused by tissue deposition of misfolded proteins that results in progressive organ damage. The most common type, immunoglobulin light chain amyloidosis (AL), is caused by clonal plasma cells that produce misfolded light chains. The purpose of this review is to provide up-to-date information on diagnosis and treatment options for AL amyloidosis. Early, accurate diagnosis is the key to effective therapy, and unequivocal identification of the amyloidogenic protein may require advanced technologies and expertise. Prognosis is dominated by the extent of cardiac involvement, and cardiac staging directs the choice of therapy. Treatment for AL amyloidosis is highly individualized, determined on the basis of age, organ dysfunction, and regimen toxicities, and should be guided by biomarkers of hematologic and cardiac response. Alkylator-based chemotherapy is effective in almost two thirds of patients. Novel agents are also active, and trials are ongoing to establish their optimal use. Treatment algorithms will continue to be refined through controlled trials. Advances in basic research have led to the identification of new drug targets and therapeutic approaches, which will be integrated with chemotherapy in the future.
Conflict of interest statement
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
Figures

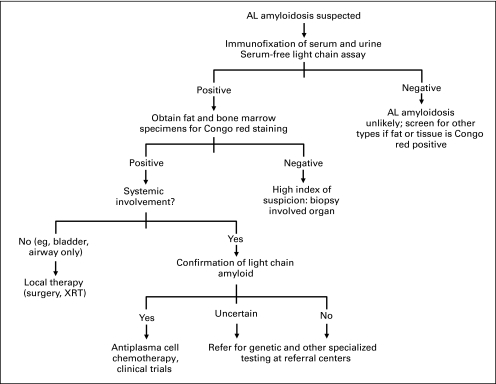
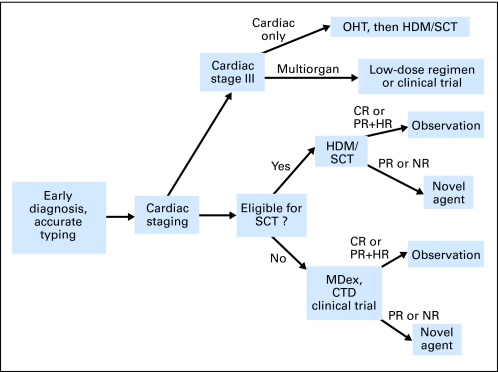
References
-
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. - PubMed
-
- Dember LM, Hawkins PN, Hazenberg BP, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. 2007;356:2349–2360. - PubMed
-
- Sekijima Y, Kelly JW, Ikeda S. Pathogenesis of and therapeutic strategies to ameliorate the transthyretin amyloidoses. Curr Pharm Des. 2008;14:3219–3230. - PubMed
-
- Pepys MB, Herbert J, Hutchinson WL, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417:254–259. - PubMed
-
- Solomon A, Weiss DT, Wall JS. Immunotherapy in systemic primary (AL) amyloidosis using amyloid-reactive monoclonal antibodies. Cancer Biother Radiopharm. 2003;18:853–860. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
