

Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis
- PMID: 21483804
- PMCID: PMC3069107
- DOI: 10.1371/journal.pgen.1001356
Widespread hypomethylation occurs early and synergizes with gene amplification during esophageal carcinogenesis
Erratum in
- PLoS Genet. 2011 May;7(5). doi: 10.1371/annotation/8dcded85-a924-40f4-a7ea-56961b87447f. Meltzer, Stephen [corrected to Meltzer, Stephen J] doi: 10.1371/annotation/8dcded85-a924-40f4-a7ea-56961b87447f
Abstract
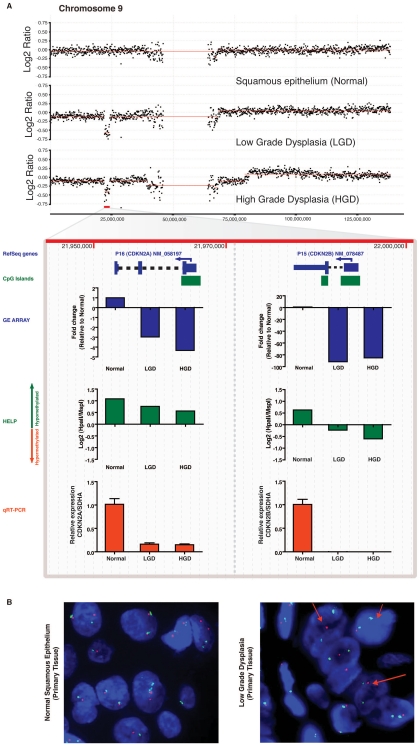
Although a combination of genomic and epigenetic alterations are implicated in the multistep transformation of normal squamous esophageal epithelium to Barrett esophagus, dysplasia, and adenocarcinoma, the combinatorial effect of these changes is unknown. By integrating genome-wide DNA methylation, copy number, and transcriptomic datasets obtained from endoscopic biopsies of neoplastic progression within the same individual, we are uniquely able to define the molecular events associated progression of Barrett esophagus. We find that the previously reported global hypomethylation phenomenon in cancer has its origins at the earliest stages of epithelial carcinogenesis. Promoter hypomethylation synergizes with gene amplification and leads to significant upregulation of a chr4q21 chemokine cluster and other transcripts during Barrett neoplasia. In contrast, gene-specific hypermethylation is observed at a restricted number of loci and, in combination with hemi-allelic deletions, leads to downregulatation of selected transcripts during multistep progression. We also observe that epigenetic regulation during epithelial carcinogenesis is not restricted to traditionally defined "CpG islands," but may also occur through a mechanism of differential methylation outside of these regions. Finally, validation of novel upregulated targets (CXCL1 and 3, GATA6, and DMBT1) in a larger independent panel of samples confirms the utility of integrative analysis in cancer biomarker discovery.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures





References
-
- Eloubeidi MA, Mason AC, Desmond RA, El-Serag HB. Temporal trends (1973-1997) in survival of patients with esophageal adenocarcinoma in the United States: a glimmer of hope? Am J Gastroenterol. 2003;98:1627–1633. - PubMed
-
- Montgomery E, Bronner MP, Goldblum JR, Greenson JK, Haber MM, et al. Reproducibility of the diagnosis of dysplasia in Barrett esophagus: a reaffirmation. Hum Pathol. 2001;32:368–378. - PubMed
-
- Montgomery E, Goldblum JR, Greenson JK, Haber MM, Lamps LW, et al. Dysplasia as a predictive marker for invasive carcinoma in Barrett esophagus: a follow-up study based on 138 cases from a diagnostic variability study. Hum Pathol. 2001;32:379–388. - PubMed
-
- Spechler SJ. Clinical practice. Barrett's Esophagus. N Engl J Med. 2002;346:836–842. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
