

Trafficking defect and proteasomal degradation contribute to the phenotype of a novel KCNH2 long QT syndrome mutation
- PMID: 21483829
- PMCID: PMC3069070
- DOI: 10.1371/journal.pone.0018273
Trafficking defect and proteasomal degradation contribute to the phenotype of a novel KCNH2 long QT syndrome mutation
Abstract
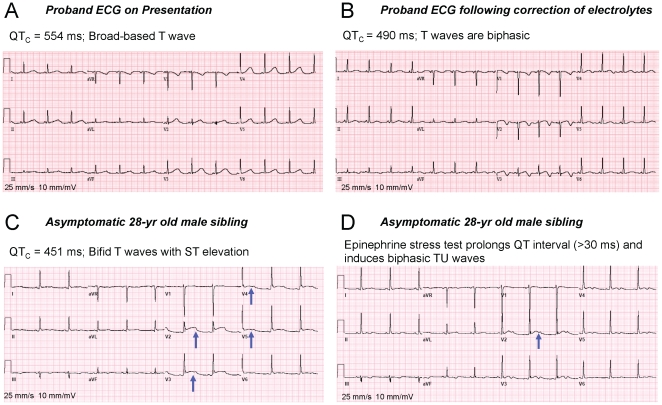
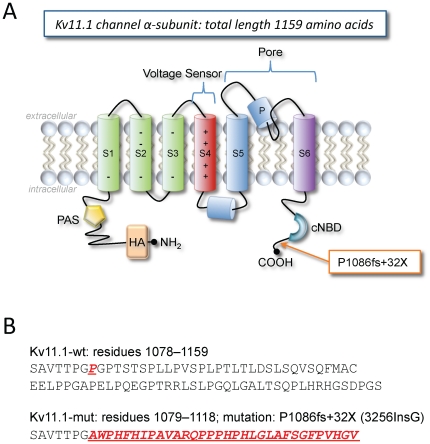
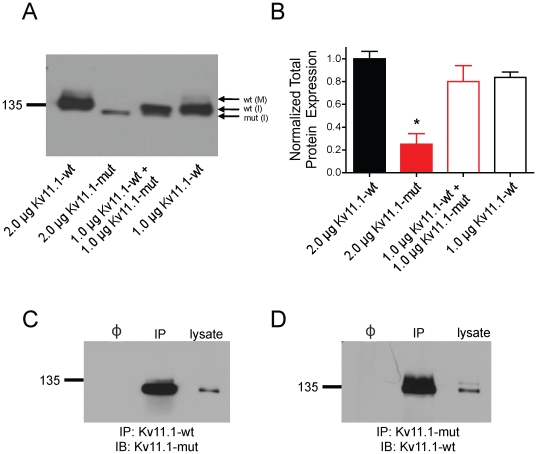
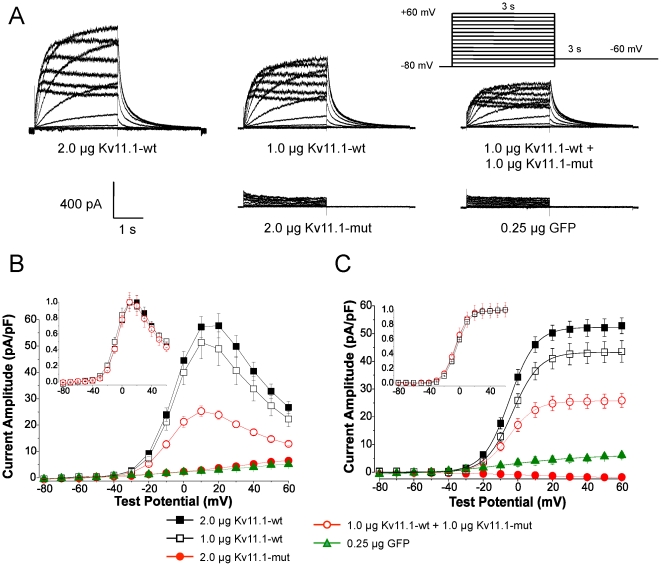
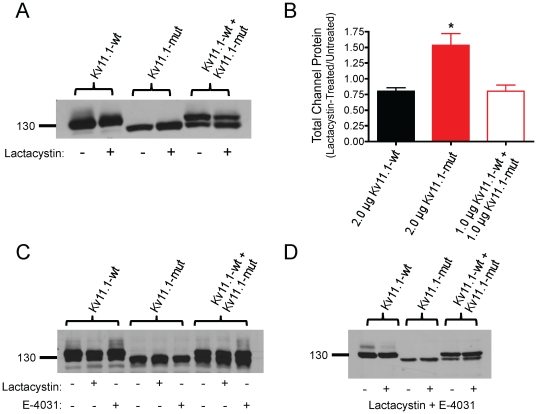
The Kv11.1 (hERG) K+ channel plays a fundamental role in cardiac repolarization. Missense mutations in KCNH2, the gene encoding Kv11.1, cause long QT syndrome (LQTS) and frequently cause channel trafficking-deficiencies. This study characterized the properties of a novel KCNH2 mutation discovered in a LQT2 patient resuscitated from a ventricular fibrillation arrest. Proband genotyping was performed by SSCP and DNA sequencing. The electrophysiological and biochemical properties of the mutant channel were investigated after expression in HEK293 cells. The proband manifested a QTc of 554 ms prior to electrolyte normalization. Mutation analysis revealed an autosomal dominant frameshift mutation at proline 1086 (P1086fs+32X; 3256InsG). Co-immunoprecipitation demonstrated that wild-type Kv11.1 and mutant channels coassemble. Western blot showed that the mutation did not produce mature complex-glycosylated Kv11.1 channels and coexpression resulted in reduced channel maturation. Electrophysiological recordings revealed mutant channel peak currents to be similar to untransfected cells. Co-expression of channels in a 1∶1 ratio demonstrated dominant negative suppression of peak Kv11.1 currents. Immunocytochemistry confirmed that mutant channels were not present at the plasma membrane. Mutant channel trafficking rescue was attempted by incubation at reduced temperature or with the pharmacological agents E-4031. These treatments did not significantly increase peak mutant currents or induce the formation of mature complex-glycosylated channels. The proteasomal inhibitor lactacystin increased the protein levels of the mutant channels demonstrating proteasomal degradation, but failed to induce mutant Kv11.1 protein trafficking. Our study demonstrates a novel dominant-negative Kv11.1 mutation, which results in degraded non-functional channels leading to a LQT2 phenotype.
Conflict of interest statement
Figures
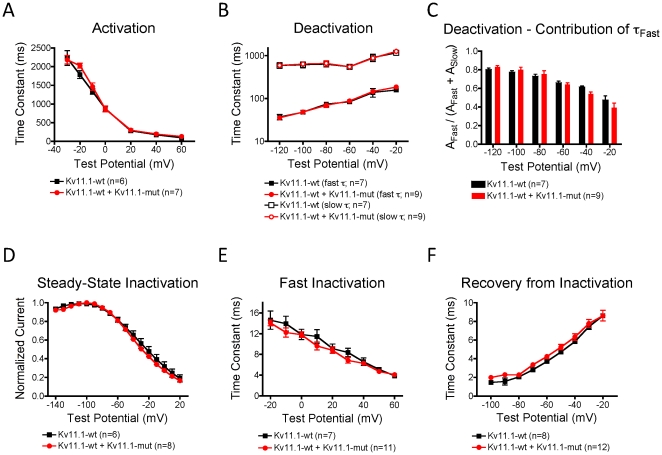
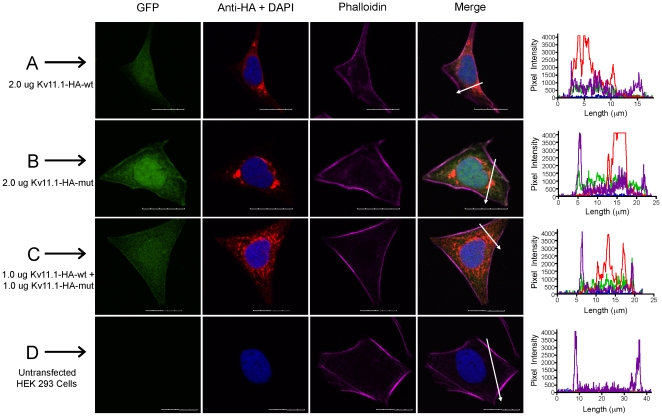
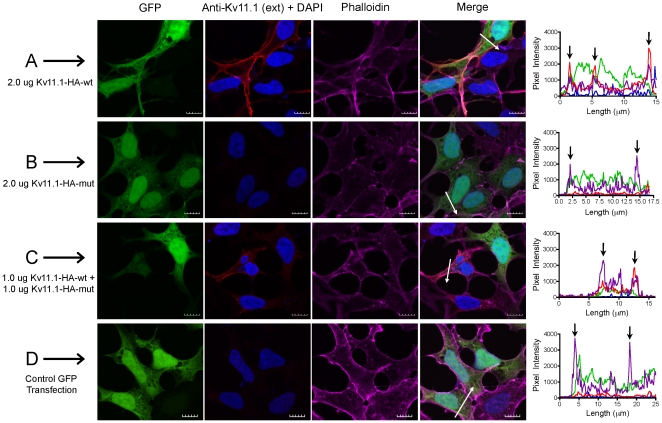
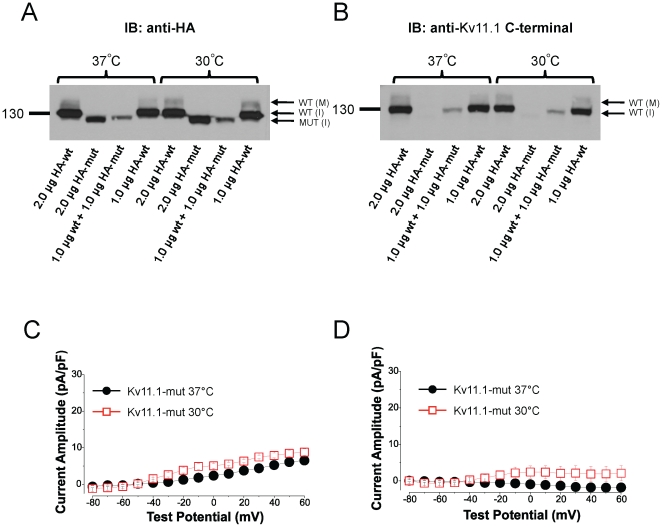
References
-
- Moss AJ. Long QT Syndrome. JAMA. 2003;289:2041–2044. - PubMed
-
- Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372:750–763. - PubMed
-
- Roden DM. Clinical practice. Long-QT syndrome. N Engl J Med. 2008;358:169–176. - PubMed
-
- Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, et al. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
