

A comprehensive benchmark study of multiple sequence alignment methods: current challenges and future perspectives
- PMID: 21483869
- PMCID: PMC3069049
- DOI: 10.1371/journal.pone.0018093
A comprehensive benchmark study of multiple sequence alignment methods: current challenges and future perspectives
Abstract
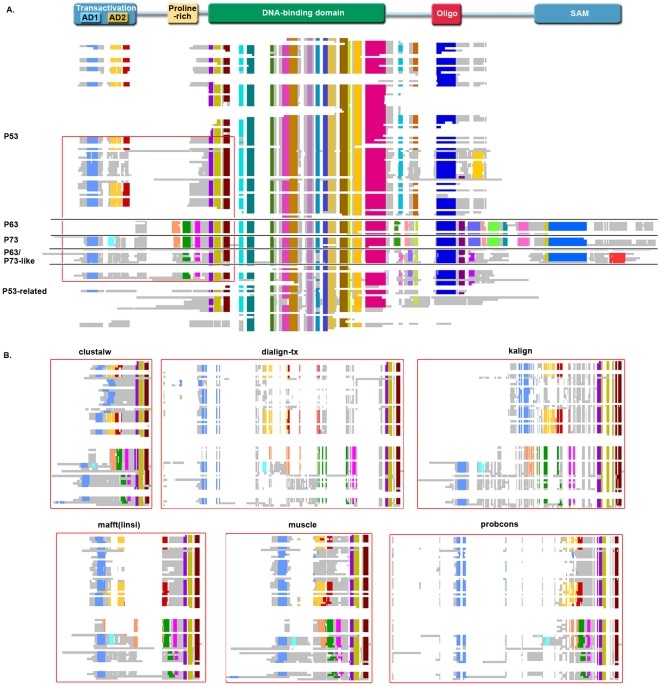
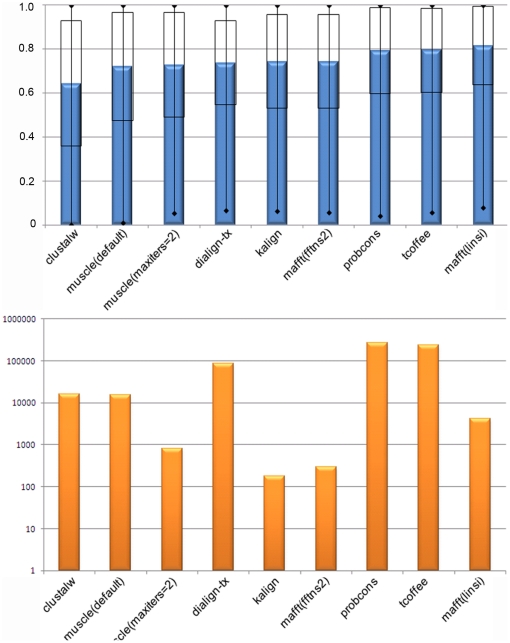
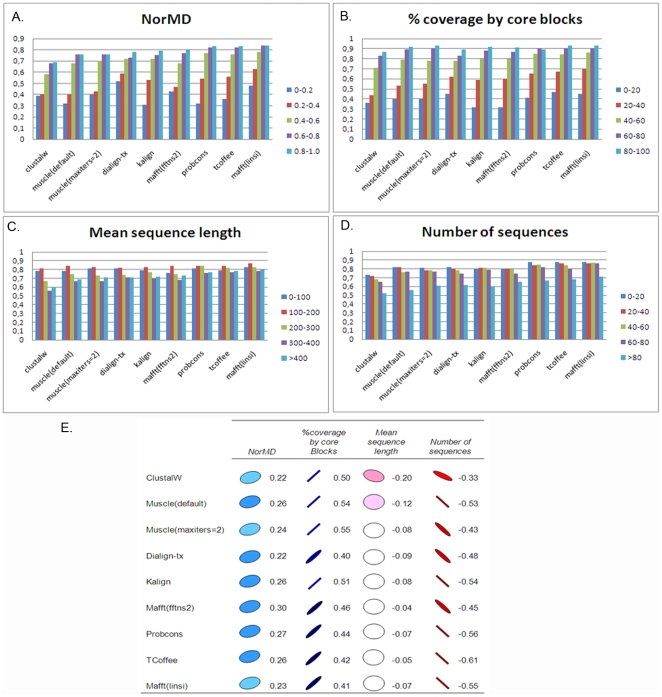
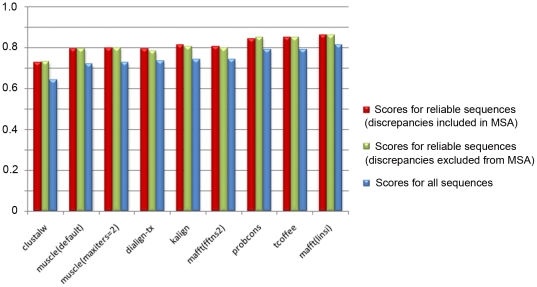
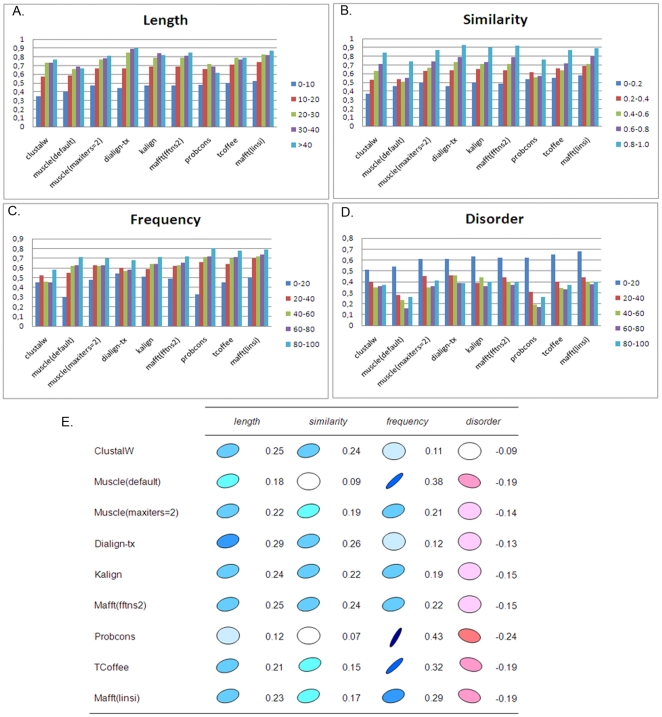
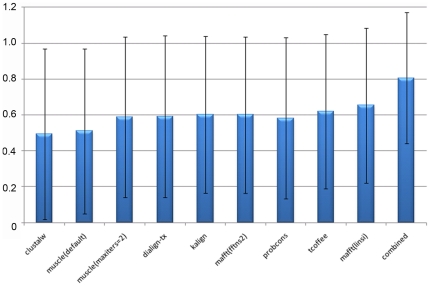
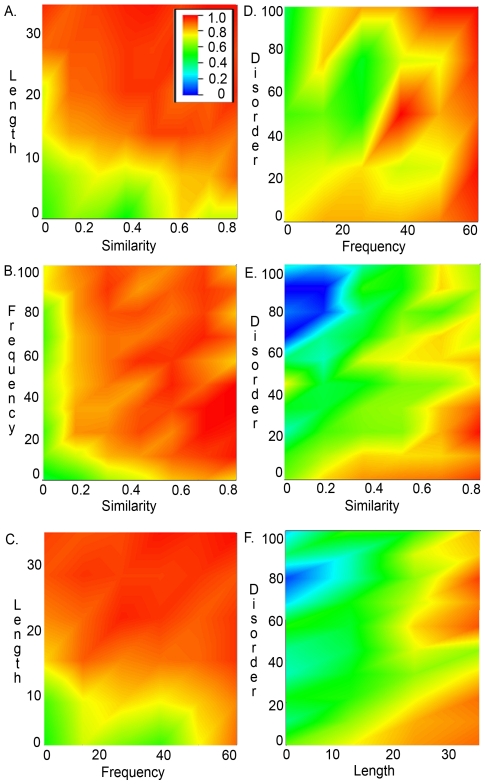
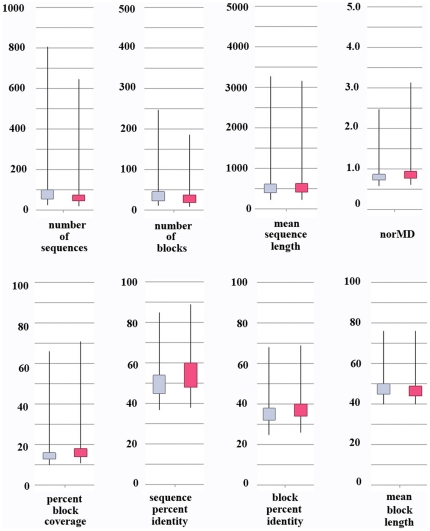
Multiple comparison or alignmentof protein sequences has become a fundamental tool in many different domains in modern molecular biology, from evolutionary studies to prediction of 2D/3D structure, molecular function and inter-molecular interactions etc. By placing the sequence in the framework of the overall family, multiple alignments can be used to identify conserved features and to highlight differences or specificities. In this paper, we describe a comprehensive evaluation of many of the most popular methods for multiple sequence alignment (MSA), based on a new benchmark test set. The benchmark is designed to represent typical problems encountered when aligning the large protein sequence sets that result from today's high throughput biotechnologies. We show that alignmentmethods have significantly progressed and can now identify most of the shared sequence features that determine the broad molecular function(s) of a protein family, even for divergent sequences. However,we have identified a number of important challenges. First, the locally conserved regions, that reflect functional specificities or that modulate a protein's function in a given cellular context,are less well aligned. Second, motifs in natively disordered regions are often misaligned. Third, the badly predicted or fragmentary protein sequences, which make up a large proportion of today's databases, lead to a significant number of alignment errors. Based on this study, we demonstrate that the existing MSA methods can be exploited in combination to improve alignment accuracy, although novel approaches will still be needed to fully explore the most difficult regions. We then propose knowledge-enabled, dynamic solutions that will hopefully pave the way to enhanced alignment construction and exploitation in future evolutionary systems biology studies.
Conflict of interest statement
Figures

References
-
- Harvey PH, Pagel MD. Oxford University Press Paris; 1991. The Comparative Method in Evolutionary Biology.
-
- Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452:745–749. - PubMed
-
- Kuipers RK, Joosten HJ, van Berkel WJ, Leferink NG, Rooijen E, et al. 3DM: systematic analysis of heterogeneous superfamily data to discover protein functionalities. Proteins. 2010;78:2101–2113. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
