

Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis
- PMID: 21487076
- PMCID: PMC3083082
- DOI: 10.1101/gr.117143.110
Exome sequencing and disease-network analysis of a single family implicate a mutation in KIF1A in hereditary spastic paraparesis
Abstract
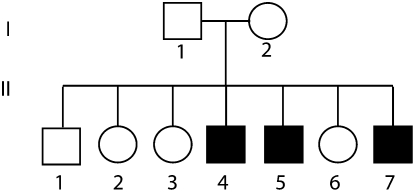
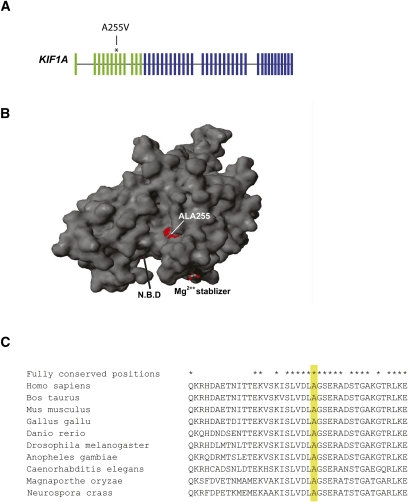
Whole exome sequencing has become a pivotal methodology for rapid and cost-effective detection of pathogenic variations in Mendelian disorders. A major challenge of this approach is determining the causative mutation from a substantial number of bystander variations that do not play any role in the disease etiology. Current strategies to analyze variations have mainly relied on genetic and functional arguments such as mode of inheritance, conservation, and loss of function prediction. Here, we demonstrate that disease-network analysis provides an additional layer of information to stratify variations even in the presence of incomplete sequencing coverage, a known limitation of exome sequencing. We studied a case of Hereditary Spastic Paraparesis (HSP) in a single inbred Palestinian family. HSP is a group of neuropathological disorders that are characterized by abnormal gait and spasticity of the lower limbs. Forty-five loci have been associated with HSP and lesions in 20 genes have been documented to induce the disorder. We used whole exome sequencing and homozygosity mapping to create a list of possible candidates. After exhausting the genetic and functional arguments, we stratified the remaining candidates according to their similarity to the previously known disease genes. Our analysis implicated the causative mutation in the motor domain of KIF1A, a gene that has not yet associated with HSP, which functions in anterograde axonal transportation. Our strategy can be useful for a large class of disorders that are characterized by locus heterogeneity, particularly when studying disorders in single families.
Figures

Comment in
-
Human disease: something old, something new.Nat Rev Genet. 2011 Jun;12(6):382-3. doi: 10.1038/nrg3007. Epub 2011 May 10. Nat Rev Genet. 2011. PMID: 21556015 No abstract available.
References
-
- Aerts S, Lambrechts D, Maity S, Van Loo P, Coessens B, De Smet F, Tranchevent LC, De Moor B, Marynen P, Hassan B, et al. 2006. Gene prioritization through genomic data fusion. Nat Biotechnol 24: 537–544 - PubMed
-
- Bittles A 2001. Consanguinity and its relevance to clinical genetics. Clin Genet 60: 89–98 - PubMed
-
- Carlson CS, Eberle MA, Rieder MJ, Smith JD, Kruglyak L, Nickerson DA 2003. Additional SNPs and linkage-disequilibrium analyses are necessary for whole-genome association studies in humans. Nat Genet 33: 518–521 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases