

An "exacerbate-reverse" strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease
- PMID: 21489983
- PMCID: PMC3129166
- DOI: 10.1074/jbc.M111.227645
An "exacerbate-reverse" strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease
Abstract
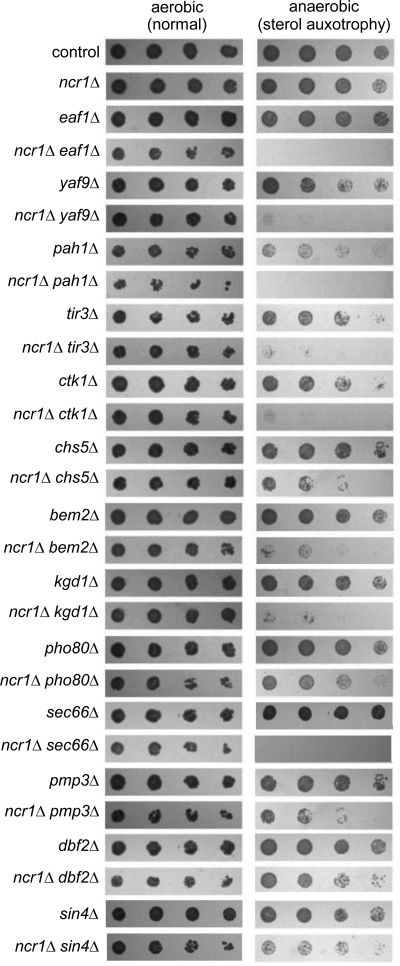
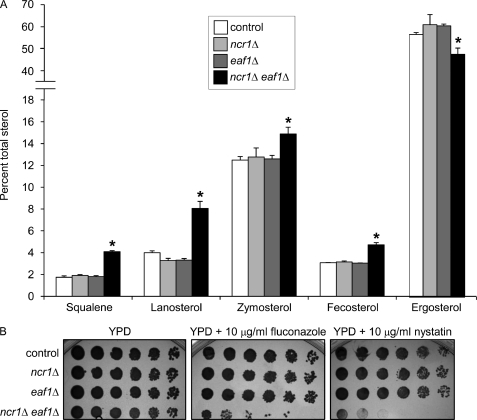
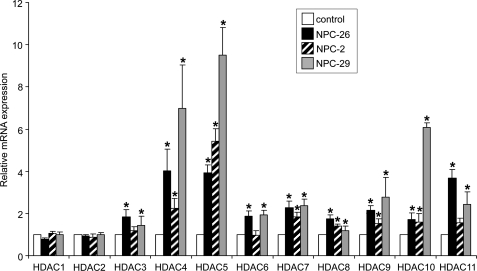
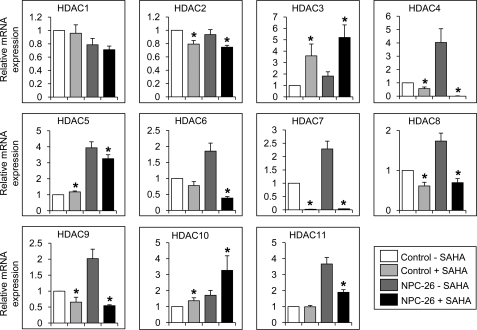
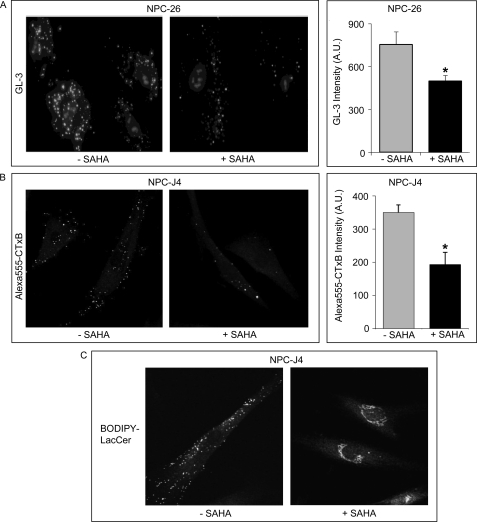
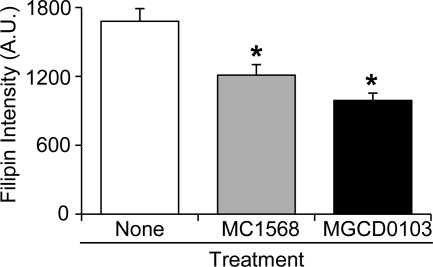
Niemann-Pick type C (NP-C) disease is a fatal lysosomal lipid storage disorder for which no effective therapy exists. A genome-wide, conditional synthetic lethality screen was performed using the yeast model of NP-C disease during anaerobiosis, an auxotrophic condition that requires yeast to utilize exogenous sterol. We identified 12 pathways and 13 genes as modifiers of the absence of the yeast NPC1 ortholog (NCR1) and quantified the impact of loss of these genes on sterol metabolism in ncr1Δ strains grown under viable aerobic conditions. Deletion of components of the yeast NuA4 histone acetyltransferase complex in ncr1Δ strains conferred anaerobic inviability and accumulation of multiple sterol intermediates. Thus, we hypothesize an imbalance in histone acetylation in human NP-C disease. Accordingly, we show that the majority of the 11 histone deacetylase (HDAC) genes are transcriptionally up-regulated in three genetically distinct fibroblast lines derived from patients with NP-C disease. A clinically approved HDAC inhibitor (suberoylanilide hydroxamic acid) reverses the dysregulation of the majority of the HDAC genes. Consequently, three key cellular diagnostic criteria of NP-C disease are dramatically ameliorated as follows: lysosomal accumulation of both cholesterol and sphingolipids and defective esterification of LDL-derived cholesterol. These data suggest HDAC inhibition as a candidate therapy for NP-C disease. We conclude that pathways that exacerbate lethality in a model organism can be reversed in human cells as a novel therapeutic strategy. This "exacerbate-reverse" approach can potentially be utilized in any model organism for any disease.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
