

FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome
- PMID: 21490157
- PMCID: PMC3725553
- DOI: 10.4049/jimmunol.1100021
FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome
Abstract
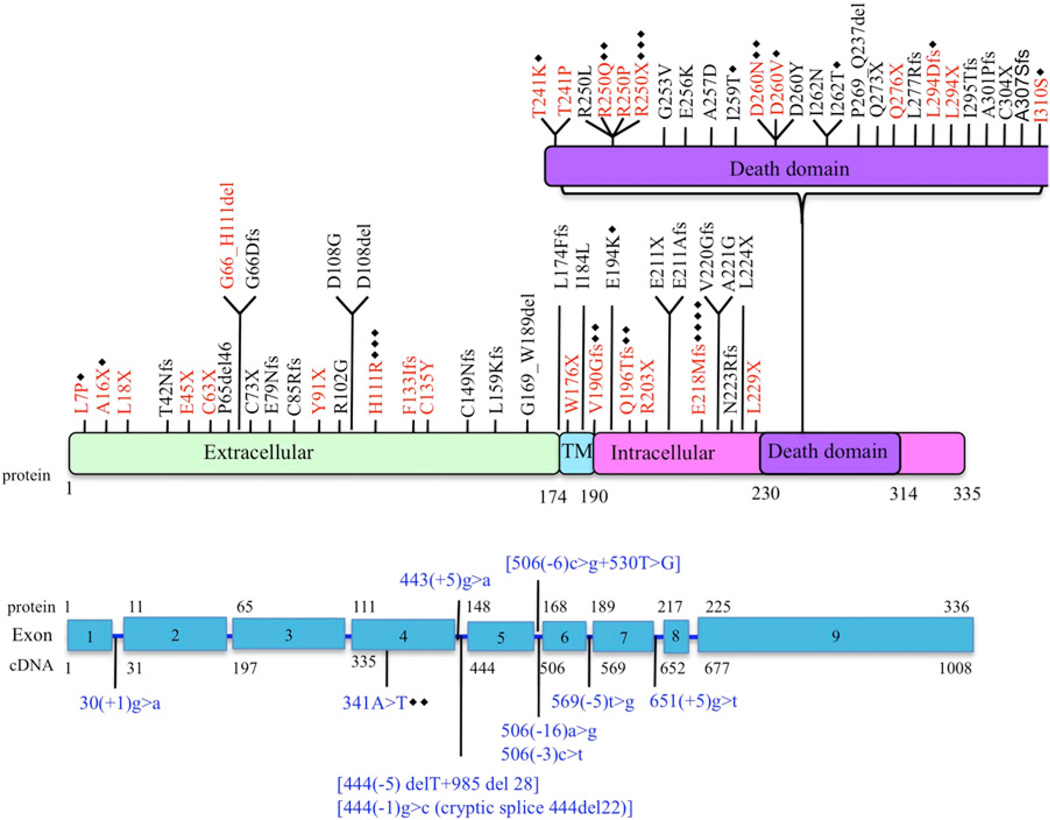
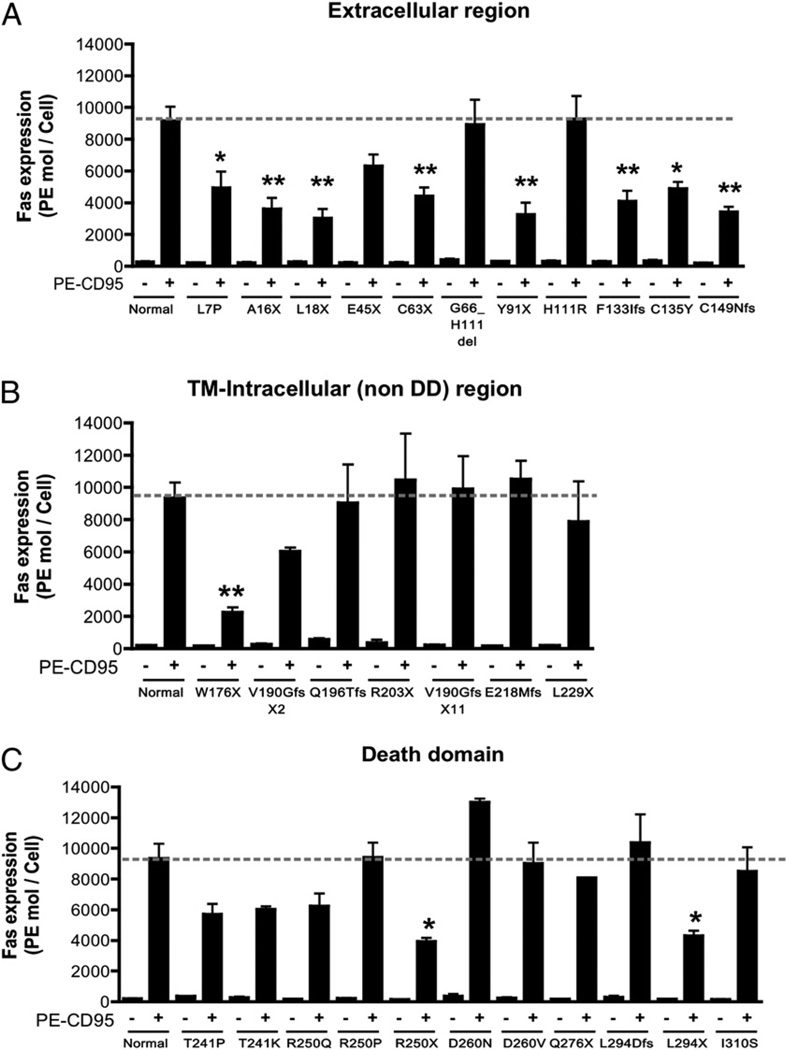
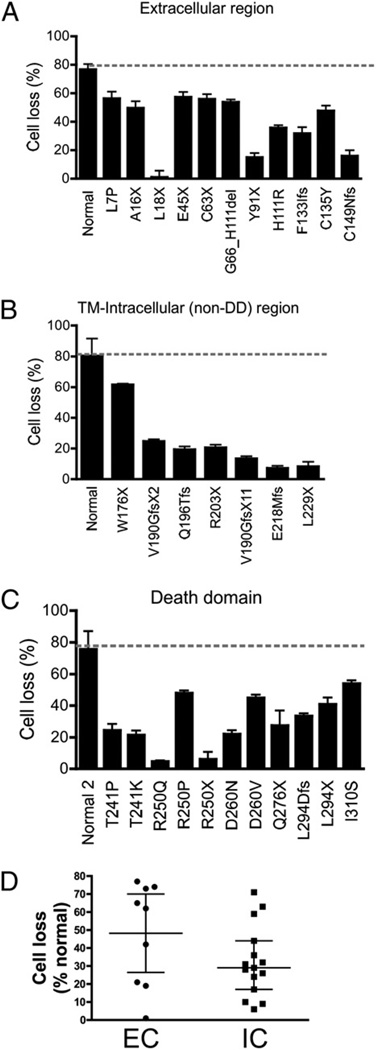
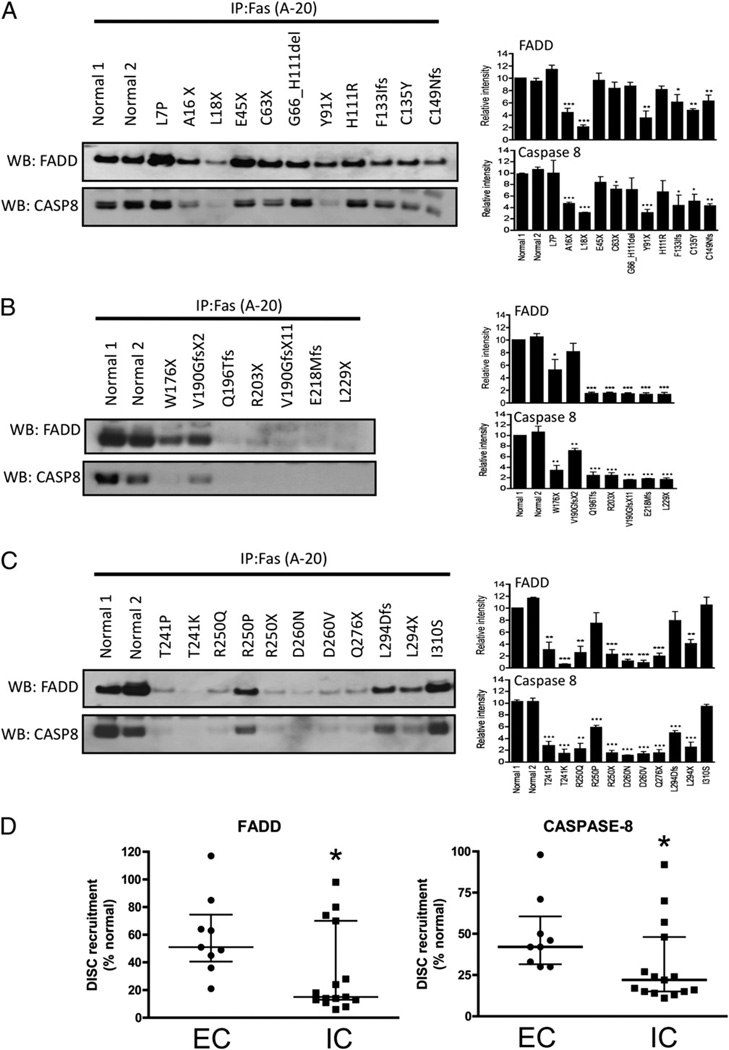
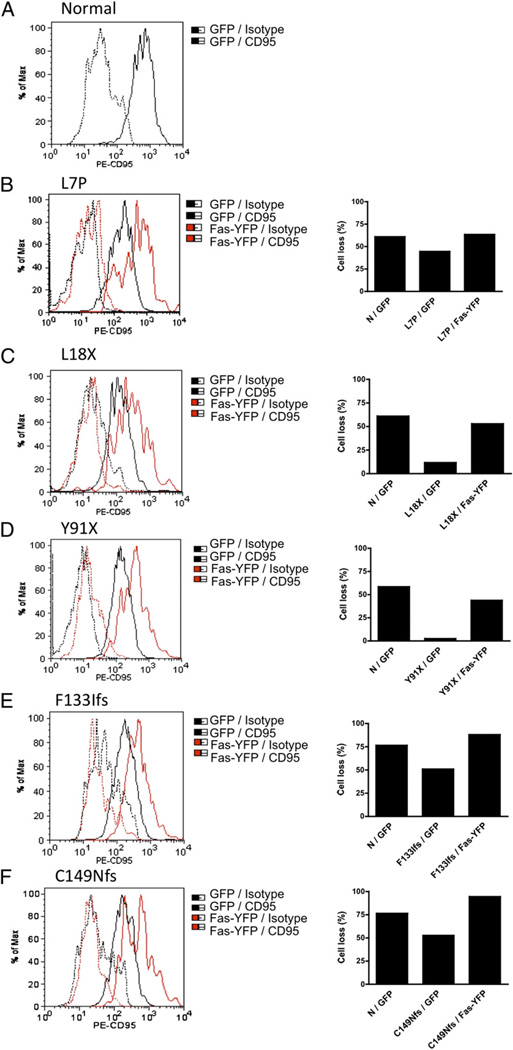
The autoimmune lymphoproliferative syndrome (ALPS) is characterized by early-onset lymphadenopathy, splenomegaly, immune cytopenias, and an increased risk for B cell lymphomas. Most ALPS patients harbor mutations in the FAS gene, which regulates lymphocyte apoptosis. These are commonly missense mutations affecting the intracellular region of the protein and have a dominant-negative effect on the signaling pathway. However, analysis of a large cohort of ALPS patients revealed that ∼30% have mutations affecting the extracellular region of FAS, and among these, 70% are nonsense, splice site, or insertions/deletions with frameshift for which no dominant-negative effect would be expected. We evaluated the latter patients to understand the mechanism(s) by which these mutations disrupted the FAS pathway and resulted in clinical disease. We demonstrated that most extracellular-region FAS mutations induce low FAS expression due to nonsense-mediated RNA decay or protein instability, resulting in defective death-inducing signaling complex formation and impaired apoptosis, although to a lesser extent as compared with intracellular mutations. The apoptosis defect could be corrected by FAS overexpression in vitro. Our findings define haploinsufficiency as a common disease mechanism in ALPS patients with extracellular FAS mutations.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures
References
-
- Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, Fleisher TA, Lim MS, Jaffe ES, Puck JM, et al. Clincal, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341–1348. - PubMed
-
- Le Deist F, Emile JF, Rieux-Laucat F, Benkerrou M, Roberts I, Brousse N, Fischer A. Clinical, immunological, and pathological consequences of Fas-deficient conditions. Lancet. 1996;348:719–723. - PubMed
-
- Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rösen-Wolff A, Peters AM, Sneller MC, Hallahan CW, Wang J, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. - PubMed
-
- Bleesing JJ, Brown MR, Straus SE, Dale JK, Siegel RM, Johnson M, Lenardo MJ, Puck JM, Fleisher TA. Immunophenotypic profiles in families with autoimmune lymphoproliferative syndrome. Blood. 2001;98:2466–2473. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
