

Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide
- PMID: 21490199
- PMCID: PMC3095121
- DOI: 10.1523/JNEUROSCI.6566-10.2011
Amyloid β-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide
Abstract
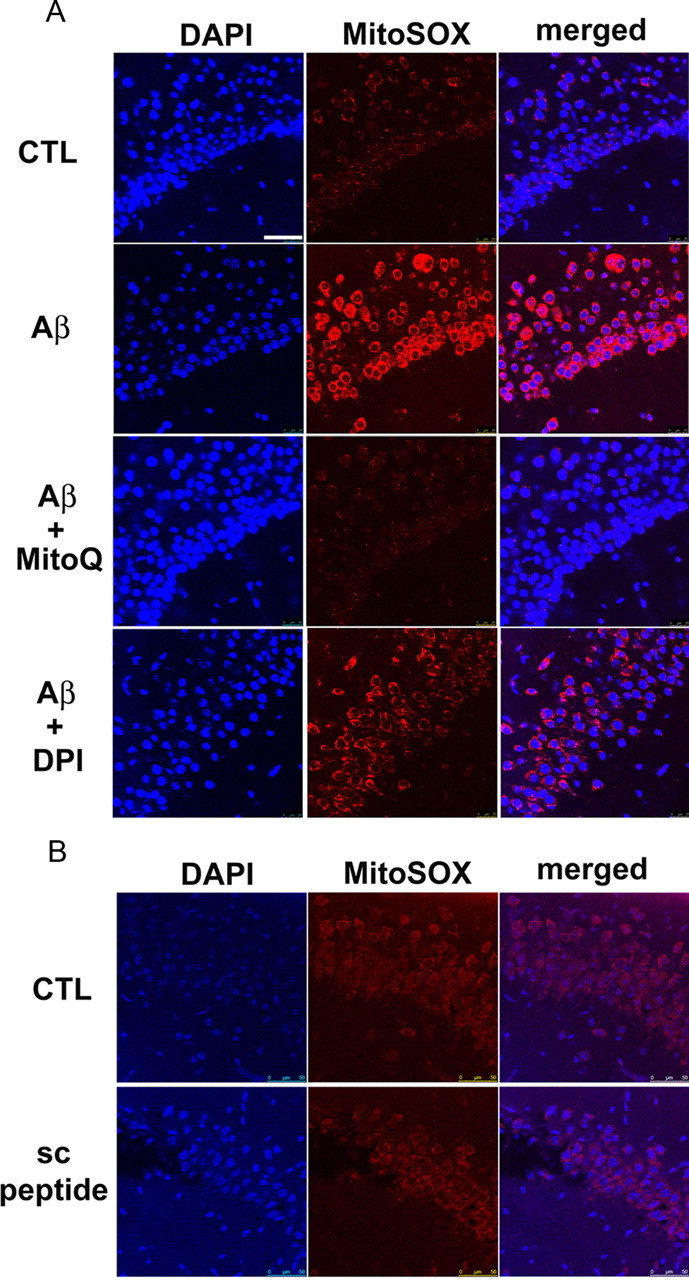
Generation of reactive oxygen species (ROS) causes cellular oxidative damage and has been implicated in the etiology of Alzheimer's disease (AD). In contrast, multiple lines of evidence indicate that ROS can normally modulate long-term potentiation (LTP), a cellular model for memory formation. We recently showed that decreasing the level of superoxide through the overexpression of mitochondrial superoxide dismutase (SOD-2) prevents memory deficits in the Tg2576 mouse model of AD. In the current study, we explored whether AD-related LTP impairments could be prevented when ROS generation from mitochondria was diminished either pharmacologically or via genetic manipulation. In wild-type hippocampal slices treated with exogenous amyloid β peptide (Aβ1-42) and in slices from APP/PS1 mutant mice that model AD, LTP was impaired. The LTP impairments were prevented by MitoQ, a mitochondria-targeted antioxidant, and EUK134, an SOD and catalase mimetic. In contrast, inhibition of NADPH oxidase either by diphenyliodonium (DPI) or by genetically deleting gp91(phox), the key enzymatic component of NADPH oxidase, had no effect on Aβ-induced LTP blockade. Moreover, live staining with MitoSOX Red, a mitochondrial superoxide indicator, combined with confocal microscopy, revealed that Aβ-induced superoxide production could be blunted by MitoQ, but not DPI, in agreement with our electrophysiological findings. Finally, in transgenic mice overexpressing SOD-2, Aβ-induced LTP impairments and superoxide generation were prevented. Our data suggest a causal relationship between mitochondrial ROS imbalance and Aβ-induced impairments in hippocampal synaptic plasticity.
Figures





Similar articles
-
Glucagon-like peptide-1 cleavage product GLP-1(9-36) amide rescues synaptic plasticity and memory deficits in Alzheimer's disease model mice.J Neurosci. 2012 Oct 3;32(40):13701-8. doi: 10.1523/JNEUROSCI.2107-12.2012. J Neurosci. 2012. PMID: 23035082 Free PMC article.
-
Beta-amyloid-mediated inhibition of NMDA receptor-dependent long-term potentiation induction involves activation of microglia and stimulation of inducible nitric oxide synthase and superoxide.J Neurosci. 2004 Jul 7;24(27):6049-56. doi: 10.1523/JNEUROSCI.0233-04.2004. J Neurosci. 2004. PMID: 15240796 Free PMC article.
-
Bis(propyl)-cognitin Prevents β-amyloid-induced Memory Deficits as Well as Synaptic Formation and Plasticity Impairments via the Activation of PI3-K Pathway.Mol Neurobiol. 2016 Aug;53(6):3832-3841. doi: 10.1007/s12035-015-9317-9. Epub 2015 Jul 10. Mol Neurobiol. 2016. PMID: 26160762
-
Cross talk between mitochondria and NADPH oxidases.Free Radic Biol Med. 2011 Oct 1;51(7):1289-301. doi: 10.1016/j.freeradbiomed.2011.06.033. Epub 2011 Jul 6. Free Radic Biol Med. 2011. PMID: 21777669 Free PMC article. Review.
-
[Effects of amyloid β-protein on hippocampal long-term potentiation].Sheng Li Xue Bao. 2010 Dec 25;62(6):479-88. Sheng Li Xue Bao. 2010. PMID: 21170492 Review. Chinese.
Cited by
-
An antioxidant specifically targeting mitochondria delays progression of Alzheimer's disease-like pathology.Aging (Albany NY). 2016 Oct 6;8(11):2713-2733. doi: 10.18632/aging.101054. Aging (Albany NY). 2016. PMID: 27750209 Free PMC article.
-
Chronic oral administration of adipoRon reverses cognitive impairments and ameliorates neuropathology in an Alzheimer's disease mouse model.Mol Psychiatry. 2021 Oct;26(10):5669-5689. doi: 10.1038/s41380-020-0701-0. Epub 2020 Mar 4. Mol Psychiatry. 2021. PMID: 32132650
-
DHA-PC and PSD-95 decrease after loss of synaptophysin and before neuronal loss in patients with Alzheimer's disease.Sci Rep. 2014 Nov 20;4:7130. doi: 10.1038/srep07130. Sci Rep. 2014. PMID: 25410733 Free PMC article.
-
Consequences of inhibiting amyloid precursor protein processing enzymes on synaptic function and plasticity.Neural Plast. 2012;2012:272374. doi: 10.1155/2012/272374. Epub 2012 Jun 26. Neural Plast. 2012. PMID: 22792491 Free PMC article. Review.
-
Nrf2 activation through the PI3K/GSK-3 axis protects neuronal cells from Aβ-mediated oxidative and metabolic damage.Alzheimers Res Ther. 2020 Jan 13;12(1):13. doi: 10.1186/s13195-019-0578-9. Alzheimers Res Ther. 2020. PMID: 31931869 Free PMC article.
References
-
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. - PubMed
-
- Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puoliväli J, Scearce-Levie K, Masliah E, Mucke L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5167–5179. - PMC - PubMed
-
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources