

Fetal hemoglobin in sickle cell anemia
- PMID: 21490337
- PMCID: PMC3139383
- DOI: 10.1182/blood-2011-03-325258
Fetal hemoglobin in sickle cell anemia
Abstract
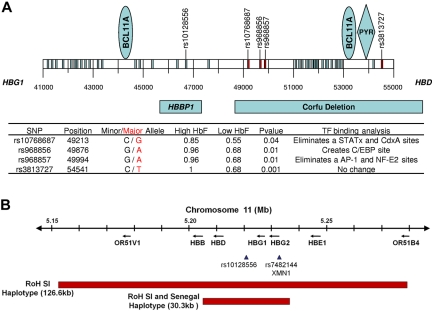
Fetal hemoglobin (HbF) is the major genetic modulator of the hematologic and clinical features of sickle cell disease, an effect mediated by its exclusion from the sickle hemoglobin polymer. Fetal hemoglobin genes are genetically regulated, and the level of HbF and its distribution among sickle erythrocytes is highly variable. Some patients with sickle cell disease have exceptionally high levels of HbF that are associated with the Senegal and Saudi-Indian haplotype of the HBB-like gene cluster; some patients with different haplotypes can have similarly high HbF. In these patients, high HbF is associated with generally milder but not asymptomatic disease. Studying these persons might provide additional insights into HbF gene regulation. HbF appears to benefit some complications of disease more than others. This might be related to the premature destruction of erythrocytes that do not contain HbF, even though the total HbF concentration is high. Recent insights into HbF regulation have spurred new efforts to induce high HbF levels in sickle cell disease beyond those achievable with the current limited repertory of HbF inducers.
Figures

References
-
- Rodgers GP, Steinberg MH. Pharmacologic treatment of sickle cell disease and thalassemia: the augmentation of fetal hemoglobin. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management (1st ed) Cambridge, United Kingdom: Cambridge University Press; 2001. pp. 1028–1051.
-
- Powars D, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63(4):921–926. - PubMed
-
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. - PubMed
-
- Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, editors. Disorders of Hemoglobin: Genetics, Pathophysiology, Clinical Management (2nd ed) Cambridge, United Kingdom: Cambridge University Press; 2009.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
