

Morphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studies
- PMID: 21490445
- PMCID: PMC3416050
- DOI: 10.1097/PAS.0b013e318212ec87
Morphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studies
Abstract
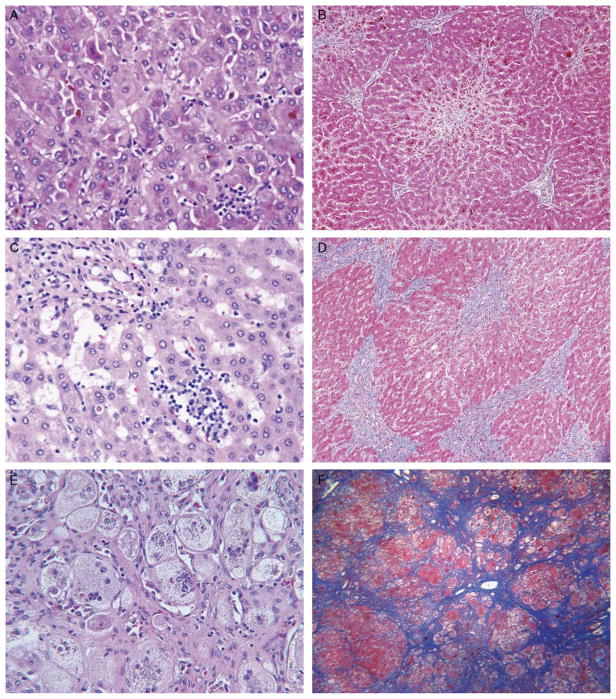
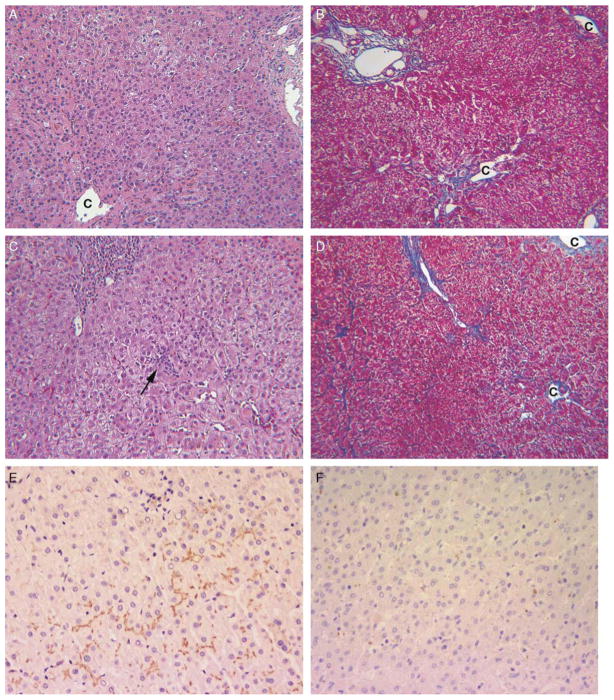
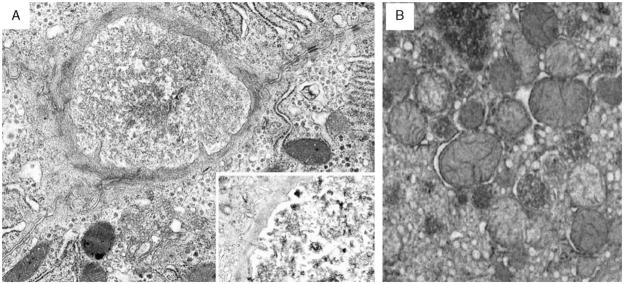
Progressive familial intrahepatic cholestasis, type 2 (PFIC2), characterized by cholestasis in infancy that may progress to cirrhosis, is caused by mutation in ABCB11, which encodes bile salt export pump (BSEP). We correlated histopathologic, immunohistochemical, and ultrastructural features in PFIC2 with specific mutations and clinical course. Twelve patients with clinical PFIC2 and ABCB11 mutations were identified, and 22 liver biopsy and explant specimens were assessed. All had hepatocellular cholestasis; most had canalicular bile plugs. At least 1 specimen from every patient had centrizonal/sinusoidal fibrosis, often with periportal fibrosis. Neonatal hepatitis-like features (inflammation, giant cells, necrosis) varied. In 2 of the 5 patients with paired specimens obtained >6 months apart, lobular and portal fibrosis worsened. Transmission electron microscopy (EM) in all 9 patients studied showed canalicular dilatation, microvilli loss, abnormal mitochondrial internal structure, and varying intracanalicular accumulation of finely granular bile. Canalicular staining for BSEP was absent in 10 patients and present in 2 patients, 1 of whom had intermittent symptoms. ABCB11 sequencing of all patients identified 6 novel and 10 previously described mutations, with nonsense, missense, and/or noncoding mutations in the 10 patients without immunohistochemically demonstrable BSEP. Missense and/or noncoding mutations were identified in the 2 patients with demonstrable BSEP, whose clinical course was more indolent. Mutations ending ABCB11 transcription appear linked, through hepatocellular necrosis and fibrosis, to worse outcome. In conclusion, light microscopy and electron microscopy findings in clinical PFIC2 can support diagnosis, but are variable and nonspecific. Therefore, no correlation between specific mutations and histopathology is yet possible.
Figures
Similar articles
-
ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history.Hepatology. 2010 May;51(5):1645-55. doi: 10.1002/hep.23539. Hepatology. 2010. PMID: 20232290
-
Relapsing features of bile salt export pump deficiency after liver transplantation in two patients with progressive familial intrahepatic cholestasis type 2.J Hepatol. 2010 Nov;53(5):981-6. doi: 10.1016/j.jhep.2010.05.025. Epub 2010 Jul 29. J Hepatol. 2010. PMID: 20800306
-
An ABCB11 variant registry and novel knockin mouse model of PFIC2 based on the clinically relevant ABCB11 E297G variant.J Lipid Res. 2025 Jul;66(7):100840. doi: 10.1016/j.jlr.2025.100840. Epub 2025 Jun 11. J Lipid Res. 2025. PMID: 40513781 Free PMC article.
-
Biosynthesis and trafficking of the bile salt export pump, BSEP: therapeutic implications of BSEP mutations.Mol Aspects Med. 2014 Jun;37:3-14. doi: 10.1016/j.mam.2013.05.001. Epub 2013 May 15. Mol Aspects Med. 2014. PMID: 23685087 Free PMC article. Review.
-
Progressive familial intrahepatic cholestasis.Clin Res Hepatol Gastroenterol. 2012 Sep;36 Suppl 1:S26-35. doi: 10.1016/S2210-7401(12)70018-9. Clin Res Hepatol Gastroenterol. 2012. PMID: 23141890 Review.
Cited by
-
Liver transplantation and the management of progressive familial intrahepatic cholestasis in children.World J Transplant. 2016 Jun 24;6(2):278-90. doi: 10.5500/wjt.v6.i2.278. World J Transplant. 2016. PMID: 27358773 Free PMC article. Review.
-
The spectrum of novel ABCB11 gene variations in children with progressive familial intrahepatic cholestasis type 2 in Pakistani cohorts.Sci Rep. 2024 Aug 14;14(1):18876. doi: 10.1038/s41598-024-59945-0. Sci Rep. 2024. PMID: 39143102 Free PMC article.
-
Vps33b is crucial for structural and functional hepatocyte polarity.J Hepatol. 2017 May;66(5):1001-1011. doi: 10.1016/j.jhep.2017.01.001. Epub 2017 Jan 9. J Hepatol. 2017. PMID: 28082148 Free PMC article.
-
Immunohistochemistry in Progressive Familial Intrahepatic Cholestasis (PFIC): Bridging Gap Between Morphology and Genetics.J Clin Exp Hepatol. 2025 Sep-Oct;15(5):102562. doi: 10.1016/j.jceh.2025.102562. Epub 2025 Mar 28. J Clin Exp Hepatol. 2025. PMID: 40384941
-
Clinical utility gene card for: progressive familial intrahepatic cholestasis type 2.Eur J Hum Genet. 2014 Apr;22(4). doi: 10.1038/ejhg.2013.187. Epub 2013 Aug 28. Eur J Hum Genet. 2014. PMID: 23982689 Free PMC article. No abstract available.
References
-
- Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2008;46:241–252. - PubMed
-
- Alonso EM, Snover DC, Montag A, et al. Histologic pathology of the liver in progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 1994;18:128–133. - PubMed
-
- Bull LN, Carlton VE, Stricker NL, et al. Genetic and morphological findings in progressive familial intrahepatic cholestasis [Byler disease (PFIC-1) and Byler syndrome]: evidence for heterogeneity. Hepatology. 1997;26:155–164. - PubMed
-
- Chen ST, Chen HL, Su YN, et al. Prenatal diagnosis of progressive familial intrahepatic cholestasis type 2. J Gastroenterol Hepatol. 2008;23:1390–1393. - PubMed
-
- Davit-Spraul A, Fabre M, Branchereau S, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51:1645–1655. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
