

The Properties of Red Blood Cells from Patients Heterozygous for HbS and HbC (HbSC Genotype)
- PMID: 21490760
- PMCID: PMC3066570
- DOI: 10.1155/2011/248527
The Properties of Red Blood Cells from Patients Heterozygous for HbS and HbC (HbSC Genotype)
Abstract
Sickle cell disease (SCD) is one of the commonest severe inherited disorders, but specific treatments are lacking and the pathophysiology remains unclear. Affected individuals account for well over 250,000 births yearly, mostly in the Tropics, the USA, and the Caribbean, also in Northern Europe as well. Incidence in the UK amounts to around 12-15,000 individuals and is increasing, with approximately 300 SCD babies born each year as well as with arrival of new immigrants. About two thirds of SCD patients are homozygous HbSS individuals. Patients heterozygous for HbS and HbC (HbSC) constitute about a third of SCD cases, making this the second most common form of SCD, with approximately 80,000 births per year worldwide. Disease in these patients shows differences from that in homozygous HbSS individuals. Their red blood cells (RBCs), containing approximately equal amounts of HbS and HbC, are also likely to show differences in properties which may contribute to disease outcome. Nevertheless, little is known about the behaviour of RBCs from HbSC heterozygotes. This paper reviews what is known about SCD in HbSC individuals and will compare the properties of their RBCs with those from homozygous HbSS patients. Important areas of similarity and potential differences will be emphasised.
Figures
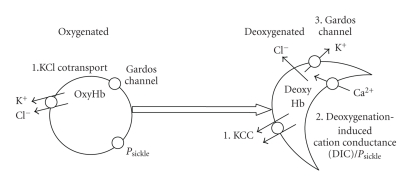
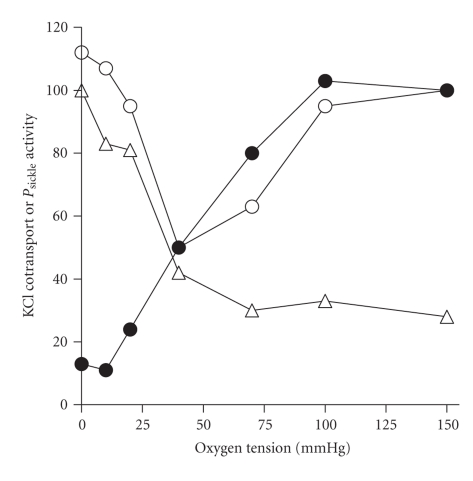
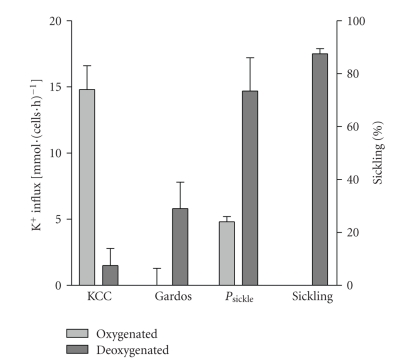
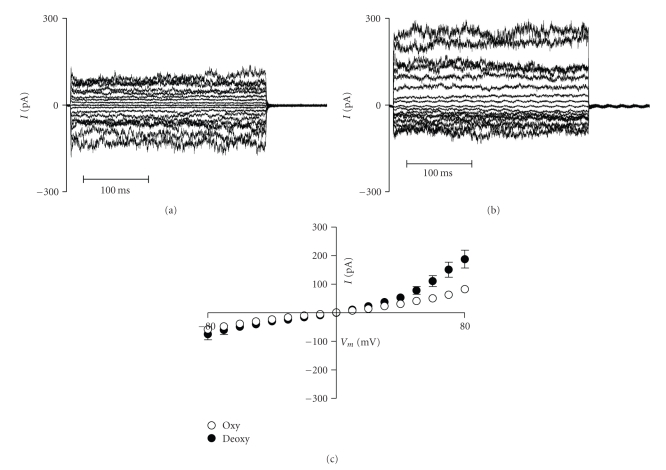
References
-
- Serjeant GR. Sickle-cell disease. The Lancet. 1997;350(9079):725–730. - PubMed
-
- Steinberg MH. Management of sickle cell disease. The New England Journal of Medicine. 1999;340(13):1021–1030. - PubMed
-
- Hickman M, Modell B, Greengross P, et al. Mapping the prevalence of sickle cell and beta thalassaemia in England: estimating and validating ethnic-specific rates. British Journal of Haematology. 1999;104(4):860–867. - PubMed
-
- NHS sickle cell & thalassaemia screening programme, 2010.
Grants and funding
LinkOut - more resources
Full Text Sources
