

Role of extracellular hemoglobin in thrombosis and vascular occlusion in patients with sickle cell anemia
- PMID: 21490767
- PMCID: PMC3065893
- DOI: 10.1155/2011/918916
Role of extracellular hemoglobin in thrombosis and vascular occlusion in patients with sickle cell anemia
Abstract
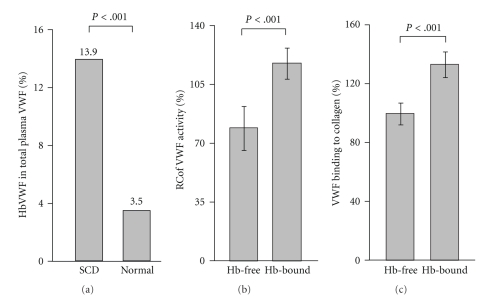
Sickle cell anemia (SCA) is a common hemolytic disorder caused by a gene mutation in the β-globin subunit of hemoglobin (Hb) and affects millions of people. The intravascular hemolysis releases excessive amount of extracellular hemoglobin (ECHb) into plasma that causes many cellular dysfunctions in patients with SCA. ECHb scavenges NO which promotes crisis events such as vasoconstriction, thrombosis and hypercoagulation. ECHb and its degradation product, heme, are known to cause oxidative damage to the vessel wall and stimulate the expression of adhesive protein ligands on vascular endothelium. Our study shows that ECHb binds potently to VWF-largest multimeric glycoprotein in circulation-through the A2-domain, and significantly inhibits its cleavage by the metalloprotease ADAMTS13. Furthermore, a subpopulation of VWF multimers bound to ECHb exists in significant amount, accounting for about 14% of total plasma VWF, in SCD patients. The Hb-bound VWF multimers are resistant to ADAMTS13, and are hyperactive in aggregating platelets. Thus, the data suggest that Hb-bound VWF multimers are ultralarge and hyperactive because they are resistant to the protease. The Hb-bound VWF multimers are elevated parallely with the level of ECHb in patients' plasma, and is associated with the pathogenesis of thrombosis and vascular occlusion in SCA.
Figures

References
-
- Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Archives of Internal Medicine. 1910;6(5):517–521.
-
- Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science. 1949;110(2865):543–548. - PubMed
-
- Ingram VM. Gene mutations in human hæmoglobin: the chemical difference between normal and sickle cell hæmoglobin. Nature. 1957;180(4581):326–328. - PubMed
-
- WHO report on sickle cell anemia and thalassemia. 2006.
-
- Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364(9442):1343–1360. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
