

Prototropic equilibria in DNA containing one-electron oxidized GC: intra-duplex vs. duplex to solvent deprotonation
- PMID: 21491657
- PMCID: PMC4677782
- DOI: 10.1039/b925496j
Prototropic equilibria in DNA containing one-electron oxidized GC: intra-duplex vs. duplex to solvent deprotonation
Abstract
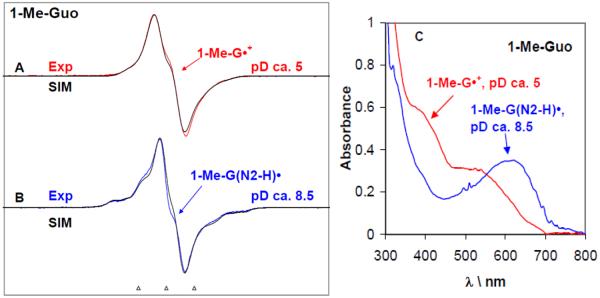
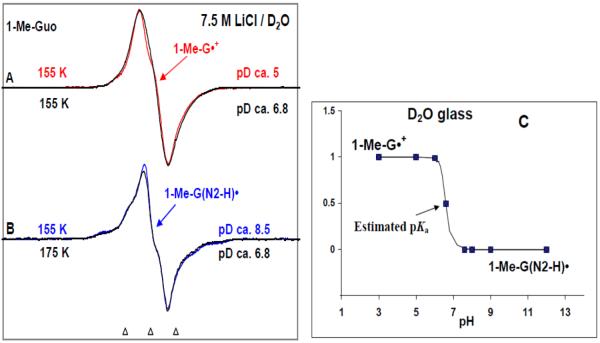
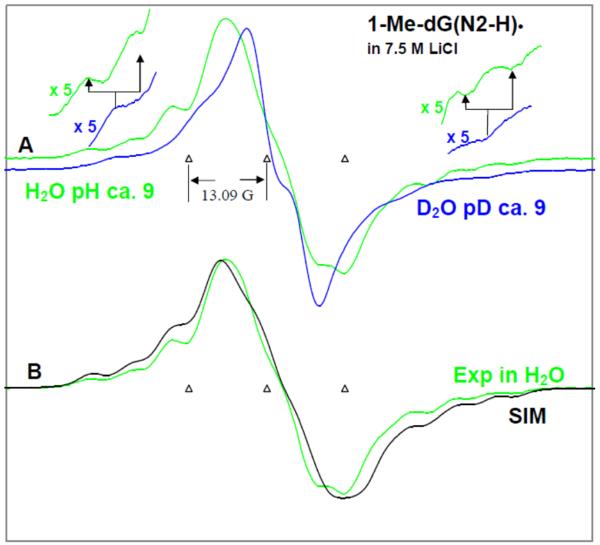
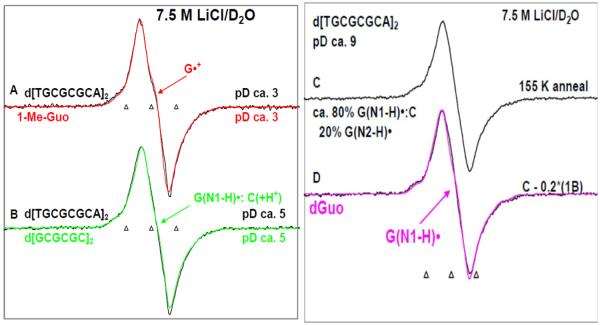
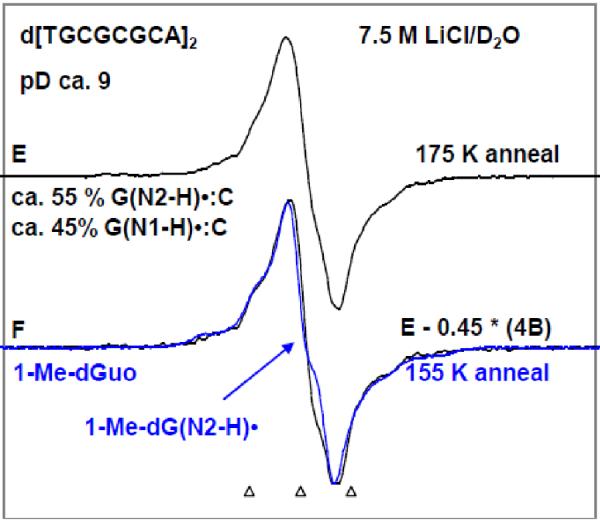
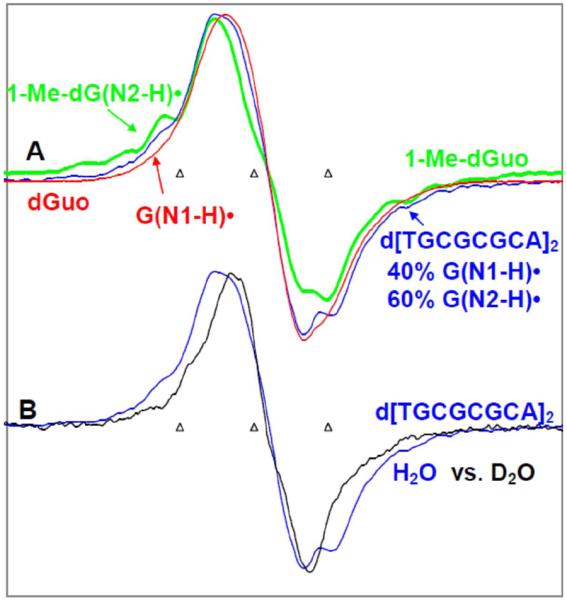
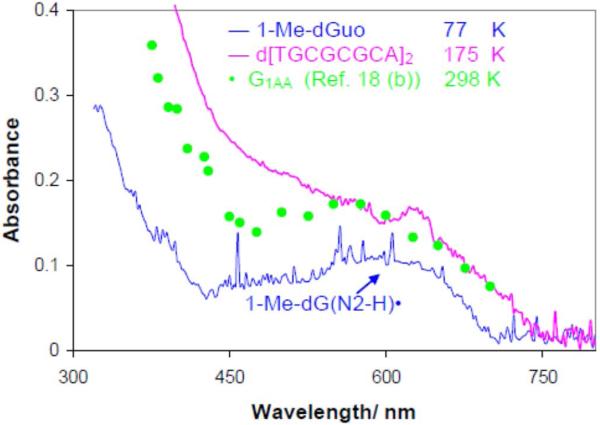
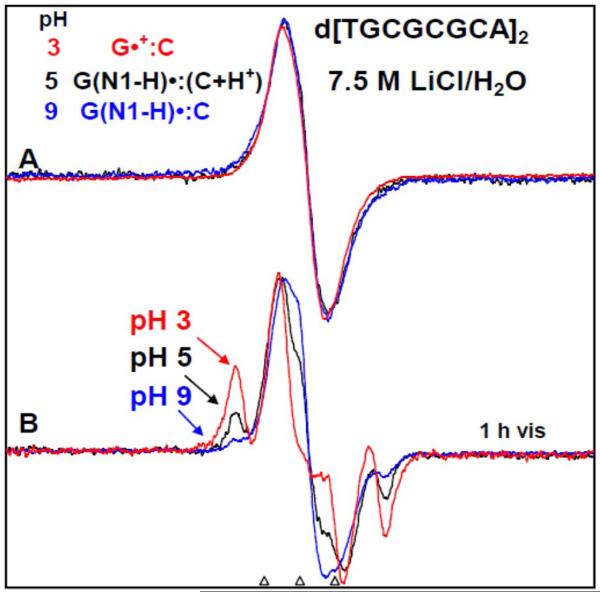
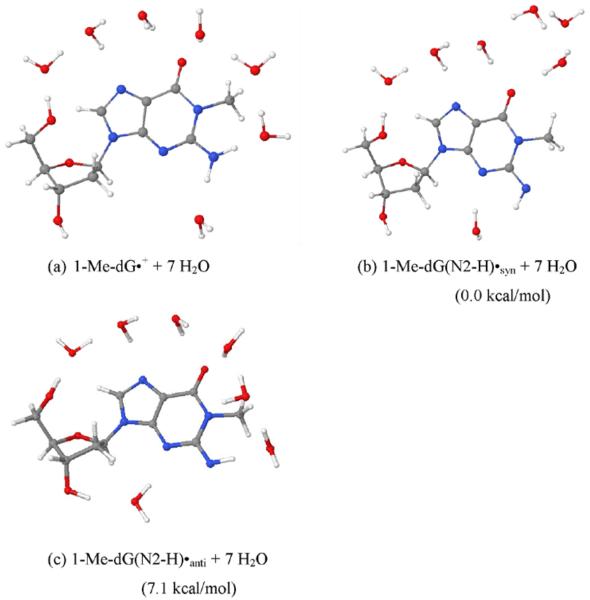
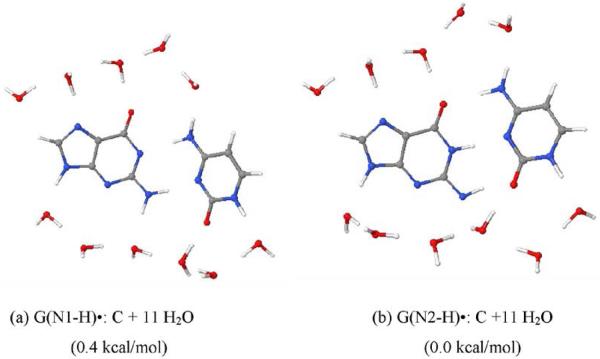
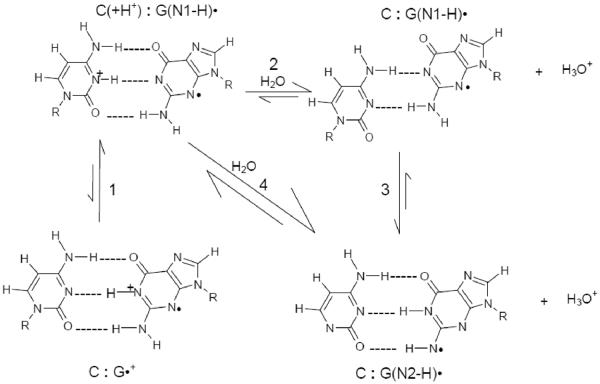
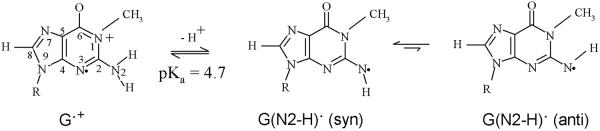
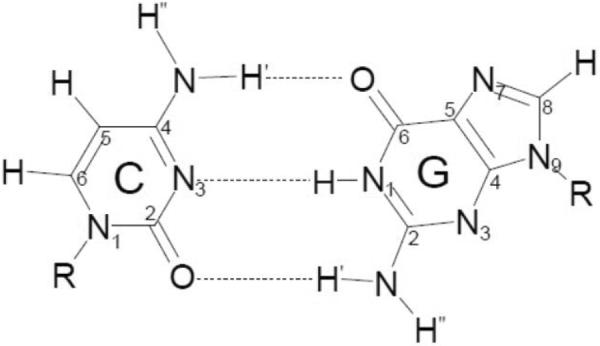
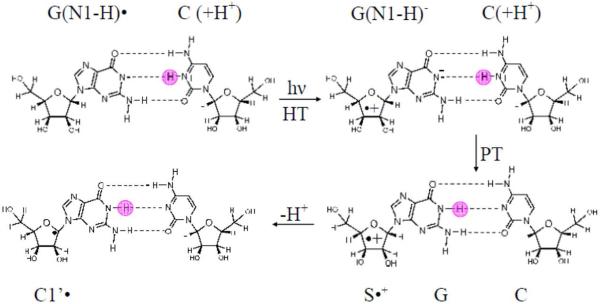
By use of ESR and UV-vis spectral studies, this work identifies the protonation states of one-electron oxidized G:C (viz. G˙+:C, G(N1–H)˙:C(+H+), G(N1–H)˙:C, and G(N2-H)˙:C) in a DNA oligomer d[TGCGCGCA]2. Benchmark ESR and UV-vis spectra from one electron oxidized 1-Me-dGuo are employed to analyze the spectral data obtained in one-electron oxidized d[TGCGCGCA]2 at various pHs. At pH ≥7, the initial site of deprotonation of one-electron oxidized d[TGCGCGCA]2 to the surrounding solvent is found to be at N1 forming G(N1–H)˙:C at 155 K. However, upon annealing to 175 K, the site of deprotonation to the solvent shifts to an equilibrium mixture of G(N1–H)˙:C and G(N2–H)˙:C. For the first time, the presence of G(N2–H)˙:C in a ds DNA-oligomer is shown to be easily distinguished from the other prototropic forms, owing to its readily observable nitrogen hyperfine coupling (Azz(N2) = 16 G). In addition, for the oligomer in H2O, an additional 8 G N2–H proton HFCC is found. This ESR identification is supported by a UV-vis absorption at 630 nm which is characteristic for G(N2–H)˙ in model compounds and oligomers. We find that the extent of photo-conversion to the C1′ sugar radical (C1′˙) in the one-electron oxidized d[TGCGCGCA]2 allows for a clear distinction among the various G:C protonation states which can not be easily distinguished by ESR or UV-vis spectroscopies with this order for the extent of photo-conversion: G˙+:C > G(N1–H)˙:C(+H+) ≫ G(N1–H)˙:C. We propose that it is the G˙+:C form that undergoes deprotonation at the sugar and this requires reprotonation of G within the lifetime of exited state
This journal is © the Owner Societies 2010
Figures
Similar articles
-
The guanine cation radical: investigation of deprotonation states by ESR and DFT.J Phys Chem B. 2006 Nov 30;110(47):24171-80. doi: 10.1021/jp064361y. J Phys Chem B. 2006. PMID: 17125389 Free PMC article.
-
Reactions of 5-methylcytosine cation radicals in DNA and model systems: thermal deprotonation from the 5-methyl group vs. excited state deprotonation from sugar.Int J Radiat Biol. 2014 Jun;90(6):433-45. doi: 10.3109/09553002.2014.884293. Epub 2014 Feb 10. Int J Radiat Biol. 2014. PMID: 24428230 Free PMC article.
-
Direct observation of the hole protonation state and hole localization site in DNA-oligomers.J Am Chem Soc. 2009 Jun 24;131(24):8614-9. doi: 10.1021/ja9014869. J Am Chem Soc. 2009. PMID: 19469533 Free PMC article.
-
The Two Faces of the Guanyl Radical: Molecular Context and Behavior.Molecules. 2021 Jun 9;26(12):3511. doi: 10.3390/molecules26123511. Molecules. 2021. PMID: 34207639 Free PMC article. Review.
-
Duplex-tetraplex equilibria in guanine- and cytosine-rich DNA.Biophys Chem. 2020 Dec;267:106473. doi: 10.1016/j.bpc.2020.106473. Epub 2020 Sep 7. Biophys Chem. 2020. PMID: 33031980 Review.
Cited by
-
How Clustered DNA Damage Can Change the Electronic Properties of ds-DNA-Differences between GAG, GAOXOG, and OXOGAOXOG.Biomolecules. 2023 Mar 11;13(3):517. doi: 10.3390/biom13030517. Biomolecules. 2023. PMID: 36979452 Free PMC article.
-
Highly oxidizing excited states of one-electron-oxidized guanine in DNA: wavelength and pH dependence.J Am Chem Soc. 2011 Mar 30;133(12):4527-37. doi: 10.1021/ja110499a. Epub 2011 Mar 7. J Am Chem Soc. 2011. PMID: 21381665 Free PMC article.
-
The Site of Azido Substitution in a Pyrimidine Nucleobase Dictates the Type of Nitrogen-Centered Radical Formed after Dissociative Electron Attachment.J Phys Chem B. 2025 Aug 14;129(32):8115-8126. doi: 10.1021/acs.jpcb.5c02751. Epub 2025 Aug 4. J Phys Chem B. 2025. PMID: 40758826 Free PMC article.
-
UV-Induced Adenine Radicals Induced in DNA A-Tracts: Spectral and Dynamical Characterization.J Phys Chem Lett. 2016 Oct 6;7(19):3949-3953. doi: 10.1021/acs.jpclett.6b01831. Epub 2016 Sep 22. J Phys Chem Lett. 2016. PMID: 27636653 Free PMC article.
-
One-electron oxidation reactions of purine and pyrimidine bases in cellular DNA.Int J Radiat Biol. 2014 Jun;90(6):423-32. doi: 10.3109/09553002.2013.877176. Epub 2014 Apr 3. Int J Radiat Biol. 2014. PMID: 24369822 Free PMC article. Review.
References
-
- von Sonntag C. Free-radical-induced DNA Damage and Its Repair. Springer-Verlag; Berlin, Heidelberg: 2006. pp. 335–447.
-
- Wagenknecht H-A, editor. Charge Transfer in DNA: From Mechanism to Application. Wiley-VCH Verlag GmbH & Co.; KGaA, Weiheim: 2005.
-
- Schuster GB, editor. Topics in Current Chemistry. Springer-Verlag; Berlin, Heidelberg: 2004. Long Range Charge Transfer in DNA. I and II.
-
- Wang Q, Fiebig T. In: Charge Migration in DNA Physics, Chemistry and Biology Perspectives. Chakraborty T, editor. Springer-Verlag; Berlin, Heidelberg: 2007. pp. 221–248.
-
- Giese B. Ann. Rev. Biochem. 2002;71:51–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous