

Combinatorial peptide ligand library treatment followed by a dual-enzyme, dual-activation approach on a nanoflow liquid chromatography/orbitrap/electron transfer dissociation system for comprehensive analysis of swine plasma proteome
- PMID: 21491903
- PMCID: PMC3125109
- DOI: 10.1021/ac200376m
Combinatorial peptide ligand library treatment followed by a dual-enzyme, dual-activation approach on a nanoflow liquid chromatography/orbitrap/electron transfer dissociation system for comprehensive analysis of swine plasma proteome
Abstract
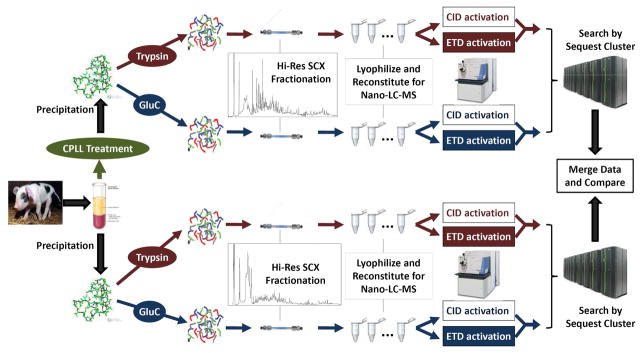
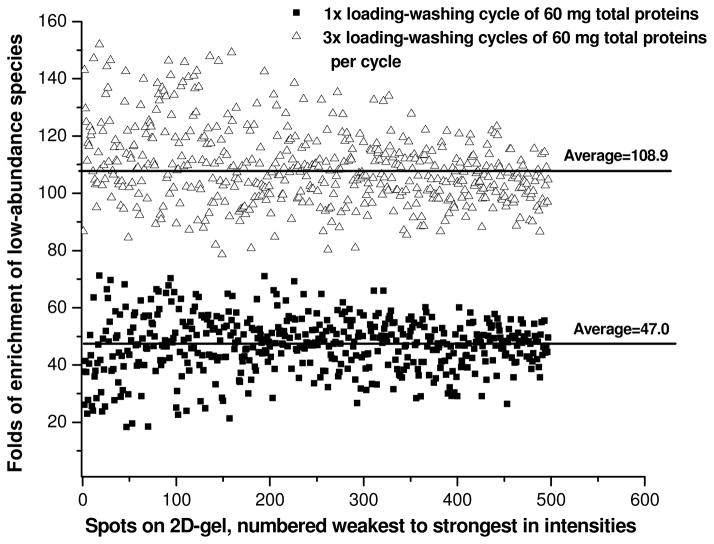
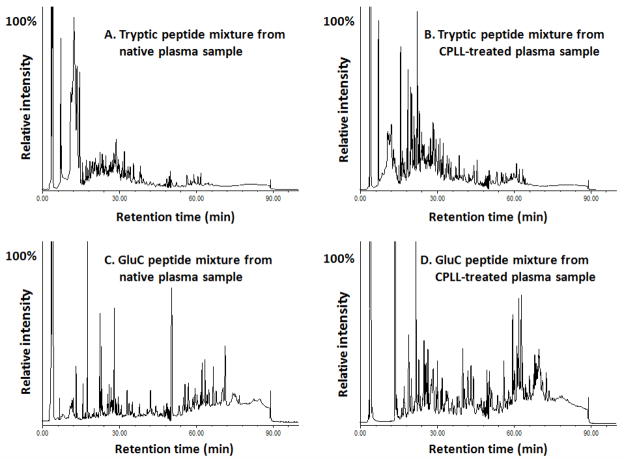
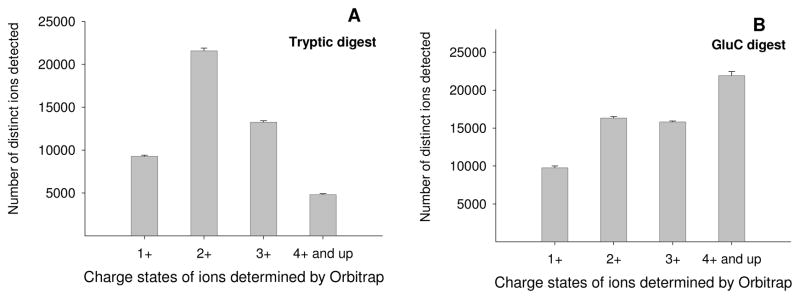
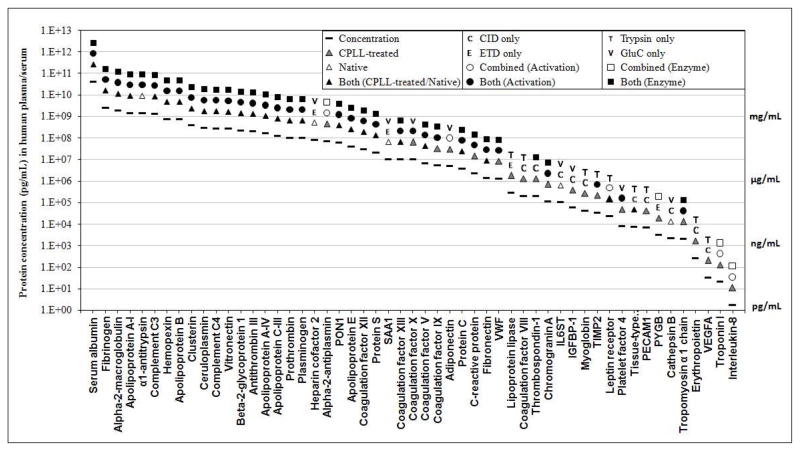
The plasma proteome holds enormous clinical potential, yet an in-depth analysis of the plasma proteome remains a daunting challenge due to its high complexity and the extremely wide dynamic range in protein concentrations. Furthermore, existing antibody-based approaches for depleting high-abundance proteins are not adaptable to the analysis of the animal plasma proteome, which is often essential for experimental pathology/pharmacology. Here we describe a highly comprehensive method for the investigation of the animal plasma proteome which employs an optimized combinatorial peptide ligand library (CPLL) treatment to reduce the protein concentration dynamic range and a dual-enzyme, dual-activation strategy to achieve high proteomic coverage. The CPLL treatment enriched the lower abundance proteins by >100-fold when the samples were loaded in moderately denaturing conditions with multiple loading-washing cycles. The native and the CPLL-treated plasma were digested in parallel by two enzymes (trypsin and GluC) carrying orthogonal specificities. By performing this differential proteolysis, the proteome coverage is improved where peptides produced by only one enzyme are poorly detectable. Digests were fractionated with high-resolution strong cation exchange chromatography and then resolved on a long, heated nano liquid chromatography column. MS analysis was performed on a linear triple quadrupole/orbitrap with two complementary activation methods (collisionally induced dissociation (CID) and electron transfer dissociation). We applied this optimized strategy to investigate the plasma proteome from swine, a prominent animal model for cardiovascular diseases (CVDs). This large-scale analysis results in identification of a total of 3421 unique proteins, spanning a concentration range of 9-10 orders of magnitude. The proteins were identified under a set of commonly accepted criteria, including a precursor mass error of <15 ppm, Xcorr cutoffs, and ≥2 unique peptides at a peptide probability of ≥95% and a protein probability of ≥99%, and the peptide false-positive rate of the data set was 1.8% as estimated by searching the reversed database. CPLL treatment resulted in 55% more identified proteins over those from native plasma; moreover, compared with using only trypsin and CID, the dual-enzyme/activation approach enabled the identification of 2.6-fold more proteins and substantially higher sequence coverage for most individual proteins. Further analysis revealed 657 proteins as significantly associated with CVDs (p < 0.05), which constitute five CVD-related pathways. This study represents the first in-depth investigation of a nonhuman plasma proteome, and the strategy developed here is adaptable to the comprehensive analysis of other highly complex proteomes.
Figures

Similar articles
-
Targeted proteomics of low-level proteins in human plasma by LC/MSn: using human growth hormone as a model system.J Proteome Res. 2002 Sep-Oct;1(5):459-65. doi: 10.1021/pr025537l. J Proteome Res. 2002. PMID: 12645918
-
Exploring the venom proteome of the western diamondback rattlesnake, Crotalus atrox, via snake venomics and combinatorial peptide ligand library approaches.J Proteome Res. 2009 Jun;8(6):3055-67. doi: 10.1021/pr900249q. J Proteome Res. 2009. PMID: 19371136
-
A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled with a long gradient nano-LC separation and Orbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome.J Proteome Res. 2009 Jun;8(6):2838-50. doi: 10.1021/pr900001t. J Proteome Res. 2009. PMID: 19290621 Free PMC article.
-
Enhanced sequence coverage of proteins in human cerebrospinal fluid using multiple enzymatic digestion and linear ion trap LC-MS/MS.Brief Funct Genomic Proteomic. 2006 Jun;5(2):144-53. doi: 10.1093/bfgp/ell026. Epub 2006 May 26. Brief Funct Genomic Proteomic. 2006. PMID: 16772279 Review.
-
The ProteoMiner and the FortyNiners: searching for gold nuggets in the proteomic arena.Mass Spectrom Rev. 2008 Nov-Dec;27(6):596-608. doi: 10.1002/mas.20178. Mass Spectrom Rev. 2008. PMID: 18481254 Review.
Cited by
-
Identification of Potential Megalin/Cubilin Substrates Using Extensive Proteomics Quantification from Kidney Megalin-Knockdown Mice.AAPS J. 2022 Oct 17;24(6):109. doi: 10.1208/s12248-022-00758-2. AAPS J. 2022. PMID: 36253507
-
Systematic assessment of survey scan and MS2-based abundance strategies for label-free quantitative proteomics using high-resolution MS data.J Proteome Res. 2014 Apr 4;13(4):2069-79. doi: 10.1021/pr401206m. Epub 2014 Mar 24. J Proteome Res. 2014. PMID: 24635752 Free PMC article.
-
Quantitative proteomic profiling of paired cancerous and normal colon epithelial cells isolated freshly from colorectal cancer patients.Proteomics Clin Appl. 2017 May;11(5-6):10.1002/prca.201600155. doi: 10.1002/prca.201600155. Epub 2017 Jan 20. Proteomics Clin Appl. 2017. PMID: 27943637 Free PMC article.
-
Proteomic methods for the study of porcine acute phase proteins - anything new to detect?Vet Res Commun. 2023 Dec;47(4):1801-1815. doi: 10.1007/s11259-023-10170-6. Epub 2023 Jul 15. Vet Res Commun. 2023. PMID: 37452983 Free PMC article. Review.
-
Cisplatin-induced ototoxicity is mediated by nitroxidative modification of cochlear proteins characterized by nitration of Lmo4.J Biol Chem. 2012 May 25;287(22):18674-86. doi: 10.1074/jbc.M111.297960. Epub 2012 Apr 9. J Biol Chem. 2012. PMID: 22493493 Free PMC article.
References
-
- Anderson NL, Anderson NG. Mol Cell Proteomics. 2002;1:845–867. - PubMed
-
- Anderson NL, Polanski M, Pieper R, Gatlin T, Tirumalai RS, Conrads TP, Veenstra TD, Adkins JN, Pounds JG, Fagan R, Lobley A. Mol Cell Proteomics. 2004;3:311–326. - PubMed
-
- Shen Y, Jacobs JM, Camp DG, 2nd, Fang R, Moore RJ, Smith RD, Xiao W, Davis RW, Tompkins RG. Anal Chem. 2004;76:1134–1144. - PubMed
-
- Pieper R, Gatlin CL, Makusky AJ, Russo PS, Schatz CR, Miller SS, Su Q, McGrath AM, Estock MA, Parmar PP, Zhao M, Huang ST, Zhou J, Wang F, Esquer-Blasco R, Anderson NL, Taylor J, Steiner S. Proteomics. 2003;3:1345–1364. - PubMed
-
- Jacobs JM, Adkins JN, Qian WJ, Liu T, Shen Y, Camp DG, 2nd, Smith RD. J Proteome Res. 2005;4:1073–1085. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
