

Update 2011: clinical and genetic issues in familial dilated cardiomyopathy
- PMID: 21492761
- PMCID: PMC3088091
- DOI: 10.1016/j.jacc.2011.01.015
Update 2011: clinical and genetic issues in familial dilated cardiomyopathy
Abstract
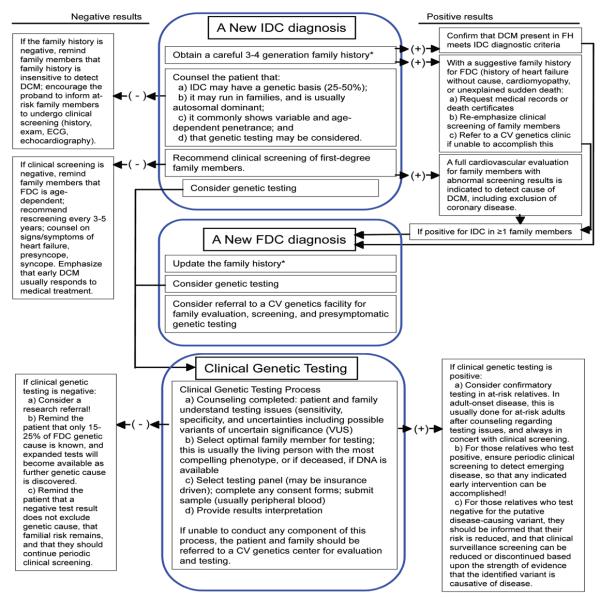
A great deal of progress has recently been made in the discovery and understanding of the genetics of familial dilated cardiomyopathy (FDC). A consensus has emerged that with a new diagnosis of idiopathic dilated cardiomyopathy (IDC), the clinical screening of first-degree family members will reveal FDC in at least 20% to 35% of those family members. Point mutations in 31 autosomal and 2 X-linked genes representing diverse gene ontogeny have been implicated in causing FDC but account for only 30% to 35% of genetic causes. Next-generation sequencing methods have dramatically decreased sequencing costs, making clinical genetic testing feasible for extensive panels of dilated cardiomyopathy genes. Next-generation sequencing also provides opportunities to discover additional genetic causes of FDC and IDC. Guidelines for evaluation and testing of FDC and IDC are now available, and when combined with FDC genetic testing and counseling, will bring FDC/IDC genetics to the forefront of cardiovascular genetic medicine.
Copyright © 2011 American College of Cardiology Foundation. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–81. - PubMed
-
- Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–16. - PubMed
-
- Richard P, Villard E, Charron P, Isnard R. The genetic bases of cardiomyopathies. J Am Coll Cardiol. 2006;48(supple A):A79–89.
-
- Ashrafian H, Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype. J Am Coll Cardiol. 2007;49:1251–64. - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
