

Ibudilast, a pharmacologic phosphodiesterase inhibitor, prevents human immunodeficiency virus-1 Tat-mediated activation of microglial cells
- PMID: 21494611
- PMCID: PMC3072977
- DOI: 10.1371/journal.pone.0018633
Ibudilast, a pharmacologic phosphodiesterase inhibitor, prevents human immunodeficiency virus-1 Tat-mediated activation of microglial cells
Abstract
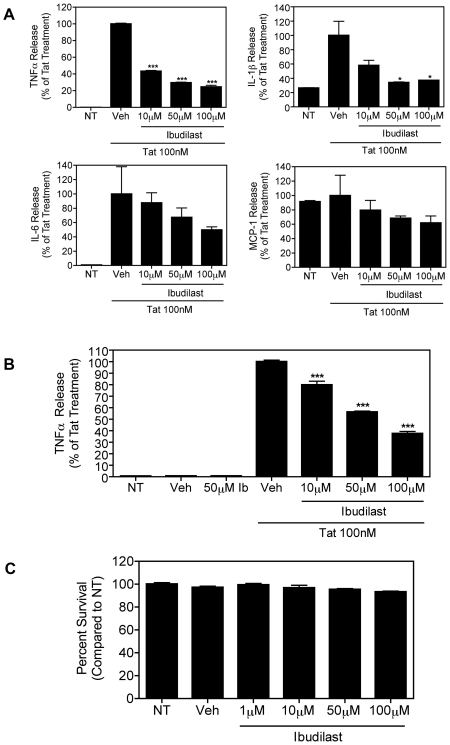
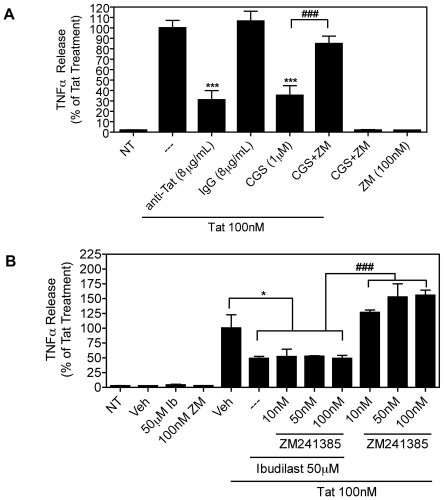
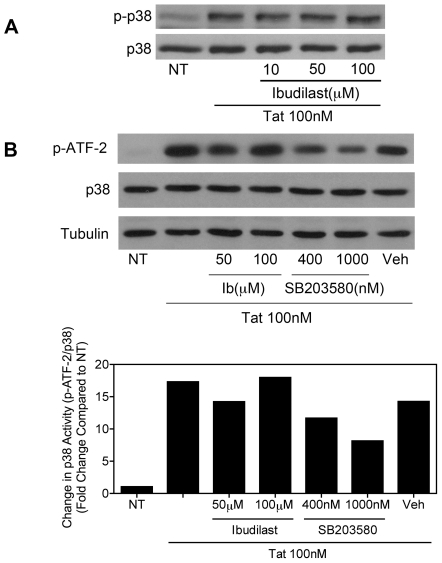
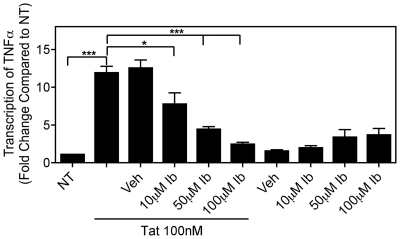
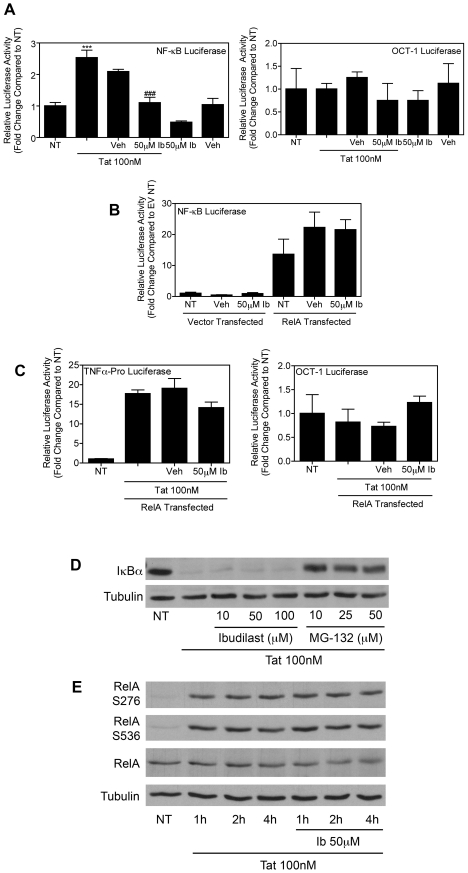
Human Immunodeficiency Virus-1 (HIV-1)-associated neurocognitive disorders (HAND) occur, in part, due to the inflammatory response to viral proteins, such as the HIV-1 transactivator of transcription (Tat), in the central nervous system (CNS). Given the need for novel adjunctive therapies for HAND, we hypothesized that ibudilast would inhibit Tat-induced excess production of pro-inflammatory cytokines, such as tumor necrosis factor-alpha (TNFα) in microglial cells. Ibudilast is a non-selective cyclic AMP phosphodiesterase inhibitor that has recently shown promise as a treatment for neuropathic pain via its ability to attenuate glial cell activation. Accordingly, here we demonstrate that pre-treatment of both human and mouse microglial cells with increasing doses of ibudilast inhibited Tat-induced synthesis of TNFα by microglial cells in a manner dependent on serine/threonine protein phosphatase activity. Ibudilast had no effect on Tat-induced p38 MAP kinase activation, and blockade of adenosine A(2A) receptor activation did not reverse ibudilast's inhibition of Tat-induced TNFα production. Interestingly, ibudilast reduced Tat-mediated transcription of TNFα, via modulation of nuclear factor-kappa B (NF-κB) signaling, as shown by transcriptional activity of NF-κB and analysis of inhibitor of kappa B alpha (IκBα) stability. Together, our findings shed light on the mechanism of ibudilast's inhibition of Tat-induced TNFα production in microglial cells and may implicate ibudilast as a potential novel adjunctive therapy for the management of HAND.
Conflict of interest statement
Figures

Similar articles
-
Ibudilast suppresses TNFalpha production by glial cells functioning mainly as type III phosphodiesterase inhibitor in the CNS.Brain Res. 1999 Aug 7;837(1-2):203-12. doi: 10.1016/s0006-8993(99)01666-2. Brain Res. 1999. PMID: 10434004
-
Nuclear factor-kappa B family member RelB inhibits human immunodeficiency virus-1 Tat-induced tumor necrosis factor-alpha production.PLoS One. 2010 Jul 29;5(7):e11875. doi: 10.1371/journal.pone.0011875. PLoS One. 2010. PMID: 20686703 Free PMC article.
-
HIV-1 TAT-mediated microglial activation: role of mitochondrial dysfunction and defective mitophagy.Autophagy. 2018;14(9):1596-1619. doi: 10.1080/15548627.2018.1476810. Epub 2018 Jul 26. Autophagy. 2018. PMID: 29966509 Free PMC article.
-
Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions.Neurochem Res. 2004 May;29(5):965-78. doi: 10.1023/b:nere.0000021241.90133.89. Neurochem Res. 2004. PMID: 15139295 Review.
-
Transactivator of Transcription (Tat)-Induced Neuroinflammation as a Key Pathway in Neuronal Dysfunction: A Scoping Review.Mol Neurobiol. 2024 Nov;61(11):9320-9346. doi: 10.1007/s12035-024-04173-w. Epub 2024 Apr 17. Mol Neurobiol. 2024. PMID: 38627350 Free PMC article.
Cited by
-
Early-life experience decreases drug-induced reinstatement of morphine CPP in adulthood via microglial-specific epigenetic programming of anti-inflammatory IL-10 expression.J Neurosci. 2011 Dec 7;31(49):17835-47. doi: 10.1523/JNEUROSCI.3297-11.2011. J Neurosci. 2011. PMID: 22159099 Free PMC article.
-
Emerging Potential of the Phosphodiesterase (PDE) Inhibitor Ibudilast for Neurodegenerative Diseases: An Update on Preclinical and Clinical Evidence.Molecules. 2022 Dec 2;27(23):8448. doi: 10.3390/molecules27238448. Molecules. 2022. PMID: 36500540 Free PMC article. Review.
-
A comprehensive study to delineate the role of an extracellular vesicle-associated microRNA-29a in chronic methamphetamine use disorder.J Extracell Vesicles. 2021 Dec;10(14):e12177. doi: 10.1002/jev2.12177. J Extracell Vesicles. 2021. PMID: 34913274 Free PMC article.
-
Discovery, synthesis, and characterization of an orally bioavailable, brain penetrant inhibitor of mixed lineage kinase 3.J Med Chem. 2013 Oct 24;56(20):8032-48. doi: 10.1021/jm401094t. Epub 2013 Oct 3. J Med Chem. 2013. PMID: 24044867 Free PMC article.
-
Interactions of HIV and drugs of abuse: the importance of glia, neural progenitors, and host genetic factors.Int Rev Neurobiol. 2014;118:231-313. doi: 10.1016/B978-0-12-801284-0.00009-9. Int Rev Neurobiol. 2014. PMID: 25175867 Free PMC article. Review.
References
-
- Resnick L, Berger JR, Shapshak P, Tourtellotte WW. Early penetration of the blood-brain-barrier by HIV. Neurology. 1988;38:9–14. - PubMed
-
- Kaul M, Lipton SA. Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res. 2006;4:307–318. - PubMed
-
- Nath A, Sacktor N. Influence of highly active antiretroviral therapy on persistence of HIV in the central nervous system. Curr Opin Neurol. 2006;19:358–361. - PubMed
-
- Tozzi V, Balestra P, Bellagamba R, Corpolongo A, Salvatori MF, et al. Persistence of neuropsychologic deficits despite long-term highly active antiretroviral therapy in patients with HIV-related neurocognitive impairment: prevalence and risk factors. J Acquir Immune Defic Syndr. 2007;45:174–182. - PubMed
-
- McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol. 2004;157:3–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
