

Genome-wide association identifies diverse causes of common variable immunodeficiency
- PMID: 21497890
- PMCID: PMC3646656
- DOI: 10.1016/j.jaci.2011.02.039
Genome-wide association identifies diverse causes of common variable immunodeficiency
Abstract
Background: Common variable immunodeficiency (CVID) is a heterogeneous immune defect characterized by hypogammaglobulinemia, failure of specific antibody production, susceptibility to infections, and an array of comorbidities.
Objective: To address the underlying immunopathogenesis of CVID and comorbidities, we conducted the first genome-wide association and gene copy number variation (CNV) study in patients with CVID.
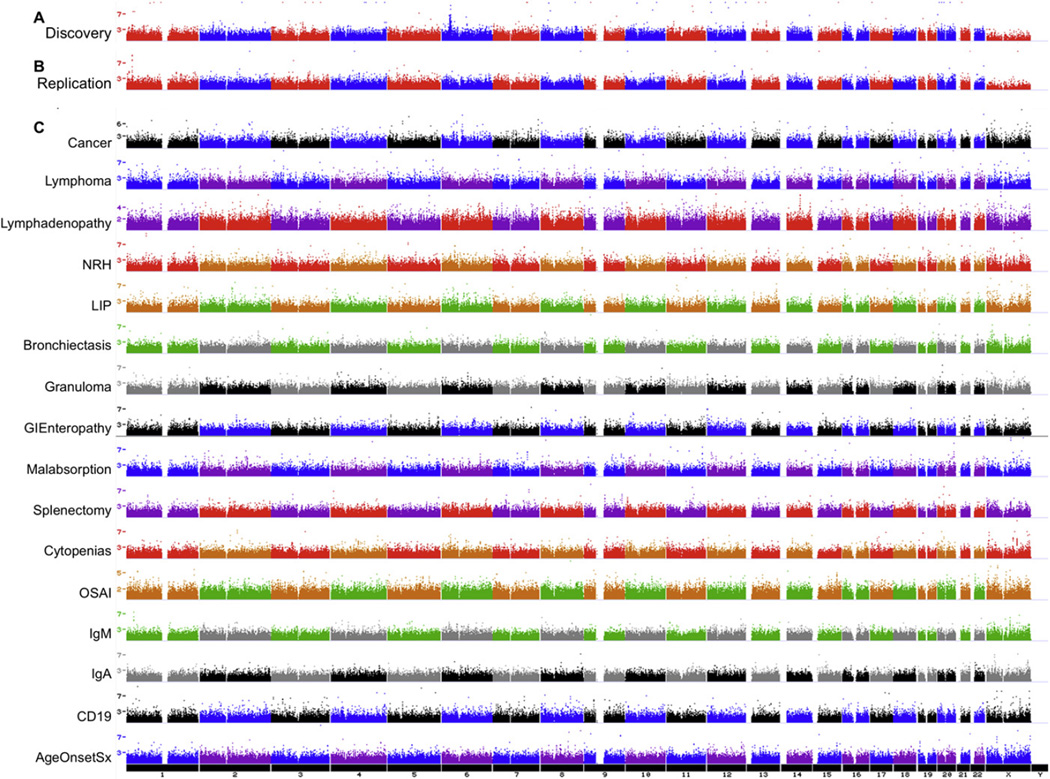
Methods: Three hundred sixty-three patients with CVID from 4 study sites were genotyped with 610,000 single nucleotide polymorphisms (SNPs). Patients were divided into a discovery cohort of 179 cases in comparison with 1,917 control subjects and a replication cohort of 109 cases and 1,114 control subjects.
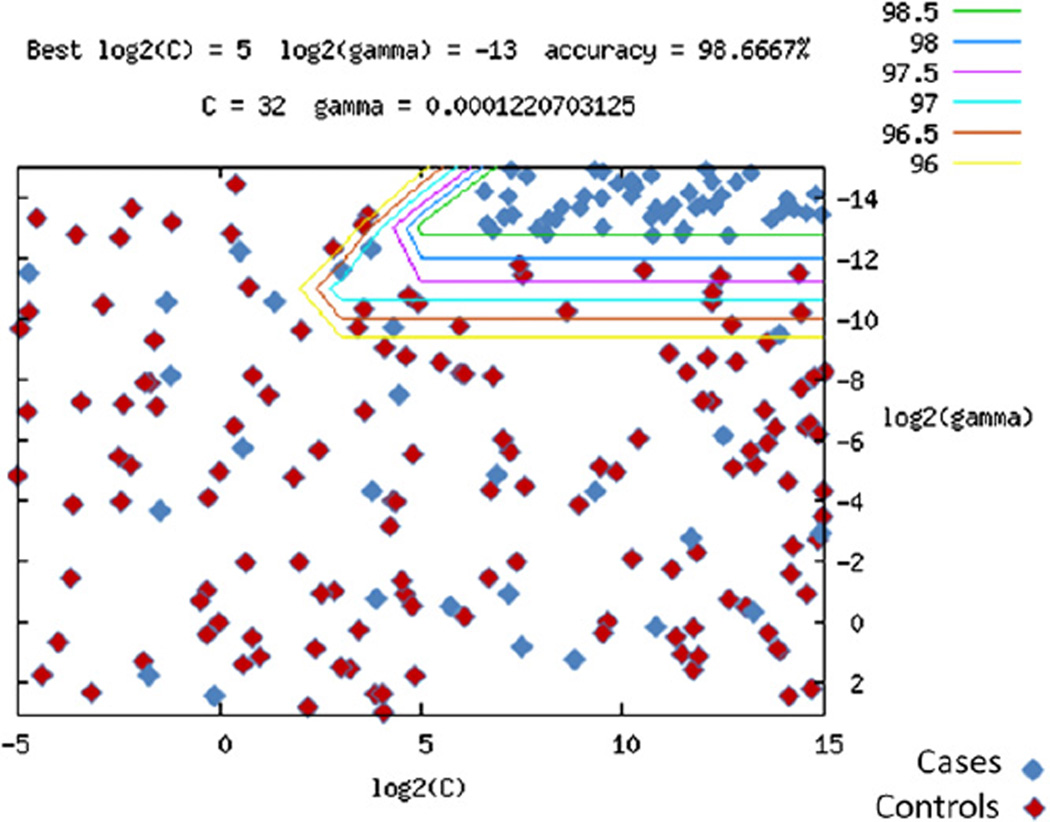
Results: Our analyses detected strong association with the MHC region and association with a disintegrin and metalloproteinase (ADAM) genes (P combined = 1.96 × 10(-7)) replicated in the independent cohort. CNV analysis defined 16 disease-associated deletions and duplications, including duplication of origin recognition complex 4L (ORC4L) that was unique to 15 cases (P = 8.66 × 10(-16)), as well as numerous unique rare intraexonic deletions and duplications suggesting multiple novel genetic causes of CVID. Furthermore, the 1,000 most significant SNPs were strongly predictive of the CVID phenotype by using a Support Vector Machine algorithm with positive and negative predictive values of 1.0 and 0.957, respectively.
Conclusion: Our integrative genome-wide analysis of SNP genotypes and CNVs has uncovered multiple novel susceptibility loci for CVID, both common and rare, which is consistent with the highly heterogeneous nature of CVID. These results provide new mechanistic insights into immunopathogenesis based on these unique genetic variations and might allow for improved diagnosis of CVID based on accurate prediction of the CVID clinical phenotypes by using our Support Vector Machine model.
Copyright © 2011 American Academy of Allergy, Asthma & Immunology. Published by Mosby, Inc. All rights reserved.
Conflict of interest statement
Disclosure of potential conflict of interest: The rest of the authors have declared that they have no conflict of interest.
Figures
References
-
- Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. - PubMed
-
- Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. - PubMed
-
- Salzer U, Chapel HM, Webster AD, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820–828. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
