

Farnesoid X receptor inhibits tamoxifen-resistant MCF-7 breast cancer cell growth through downregulation of HER2 expression
- PMID: 21499302
- PMCID: PMC4482257
- DOI: 10.1038/onc.2011.124
Farnesoid X receptor inhibits tamoxifen-resistant MCF-7 breast cancer cell growth through downregulation of HER2 expression
Abstract
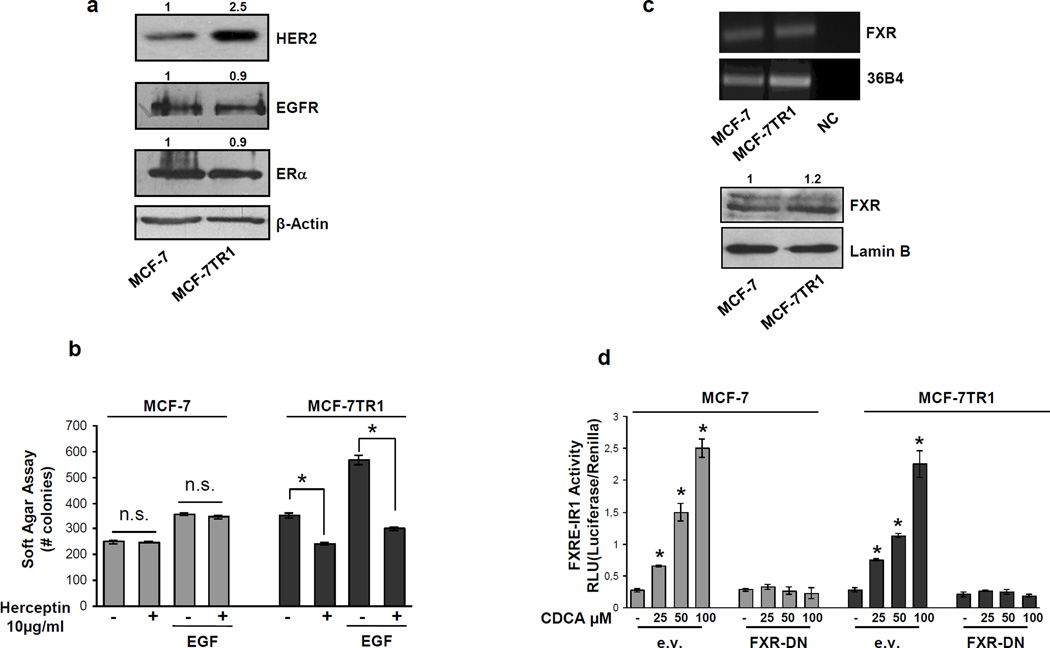
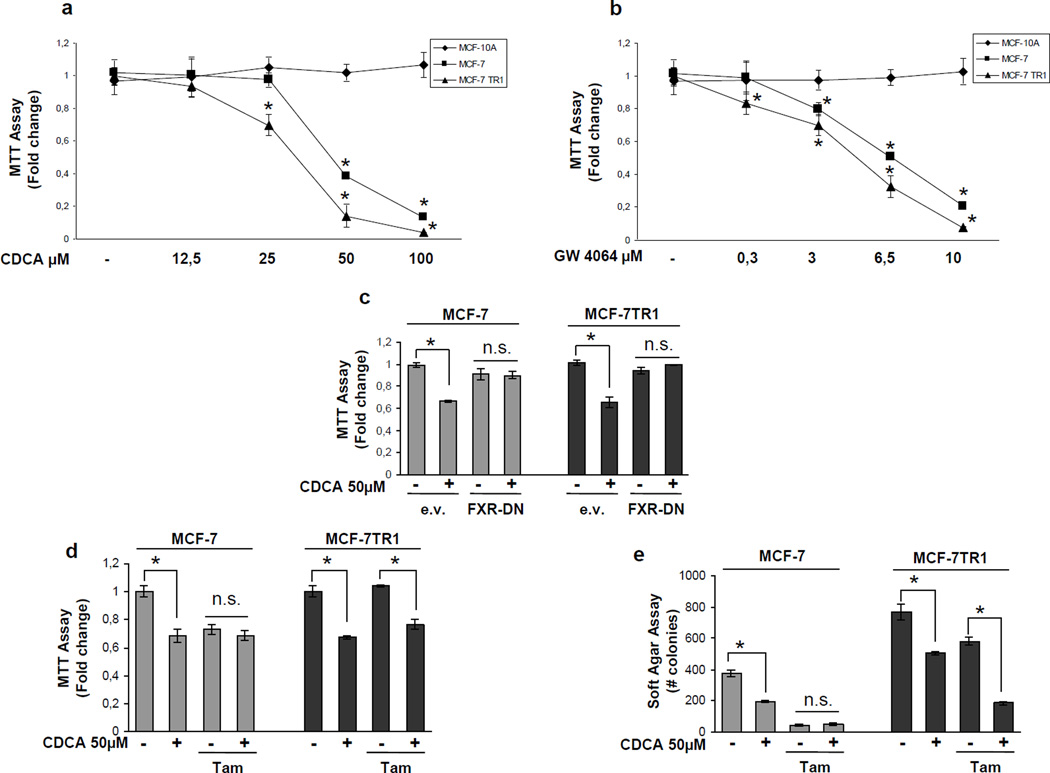
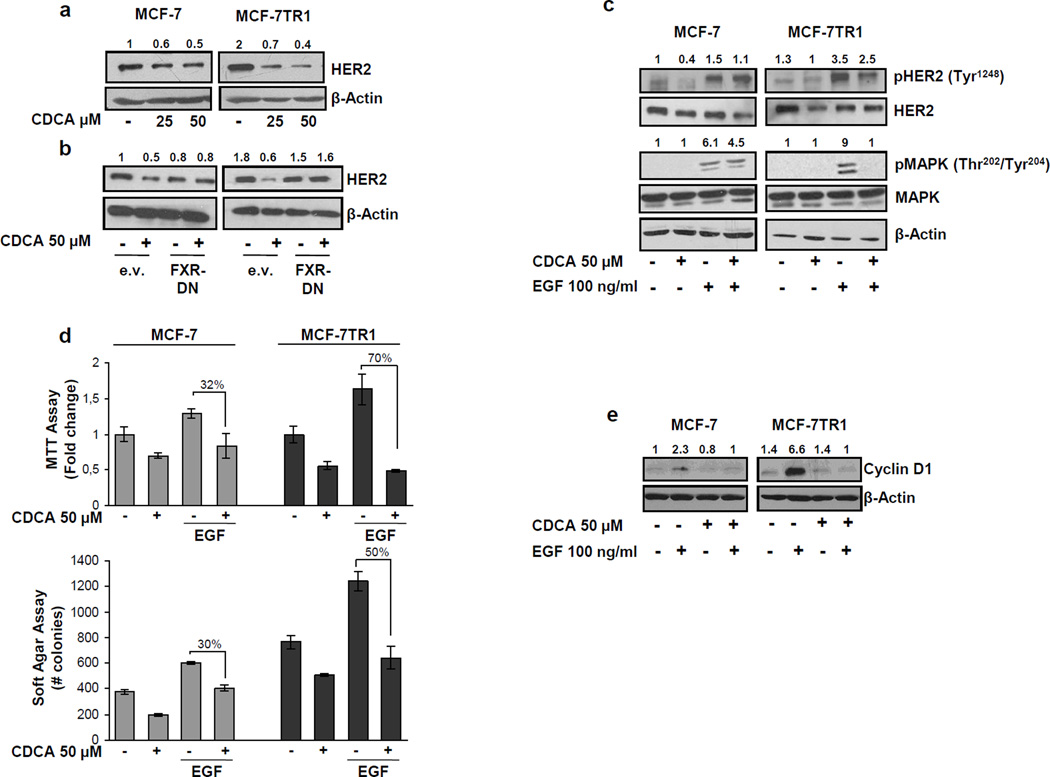
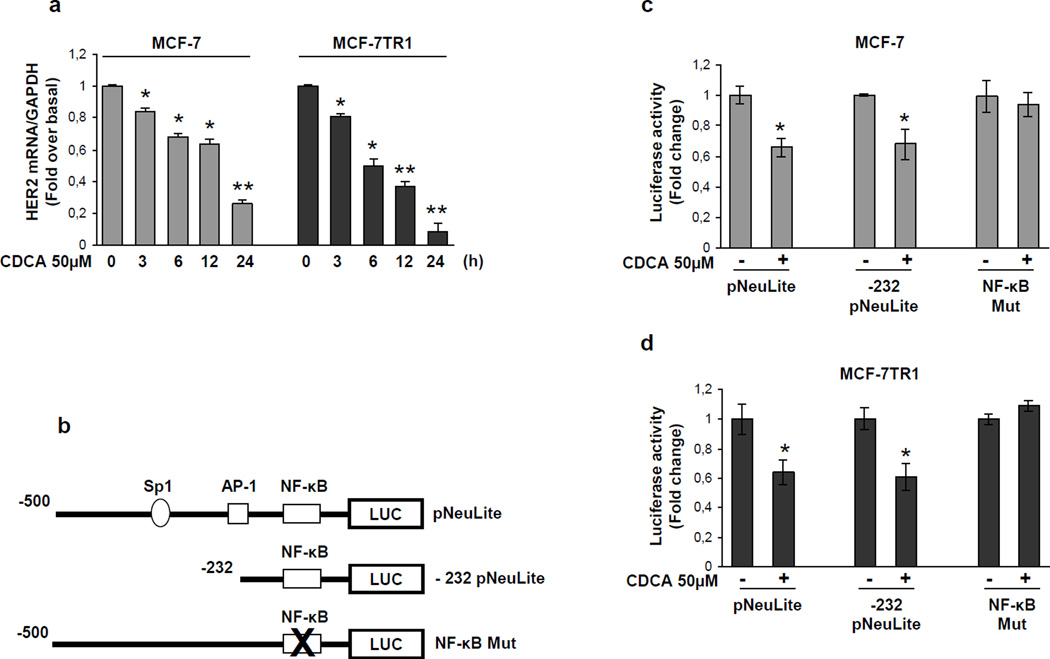
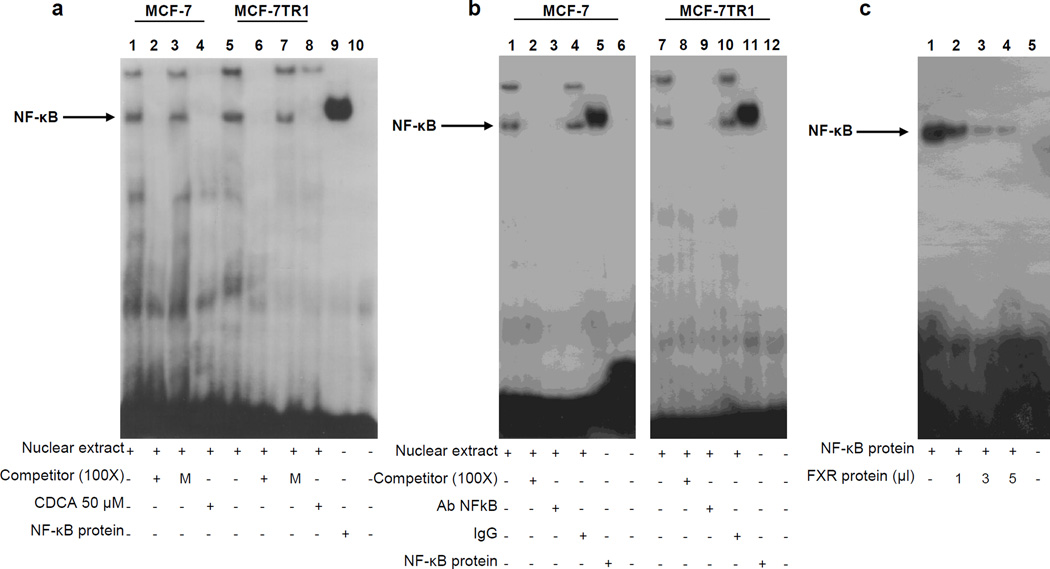
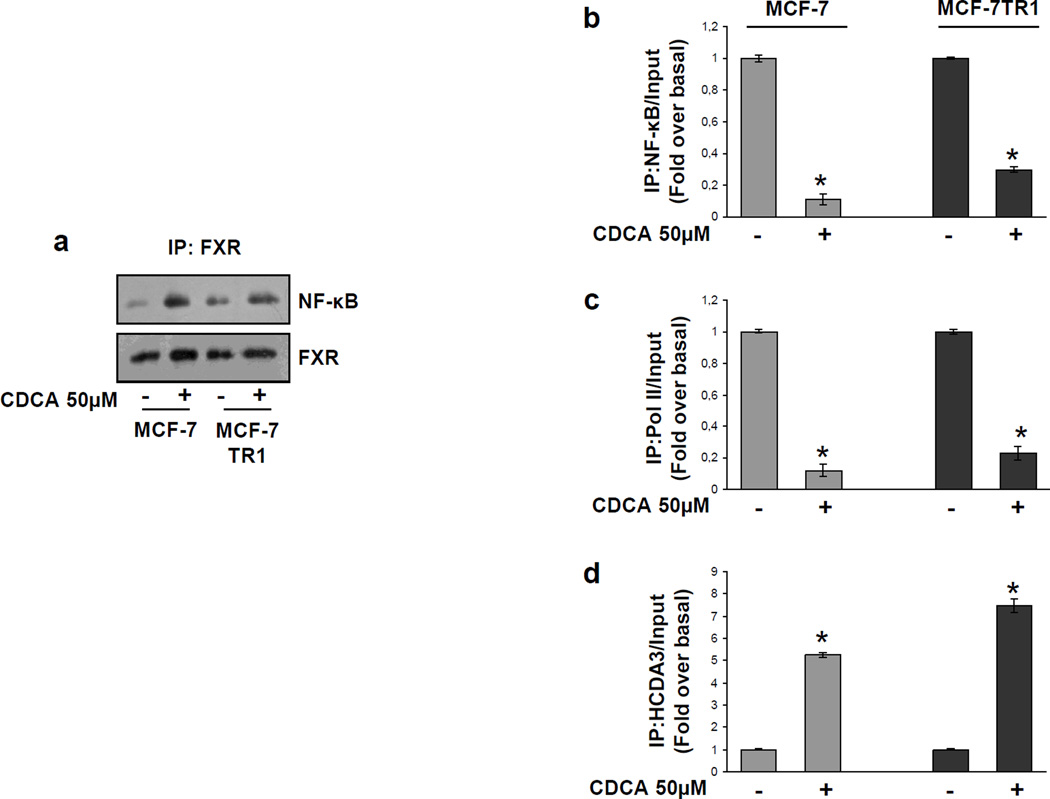
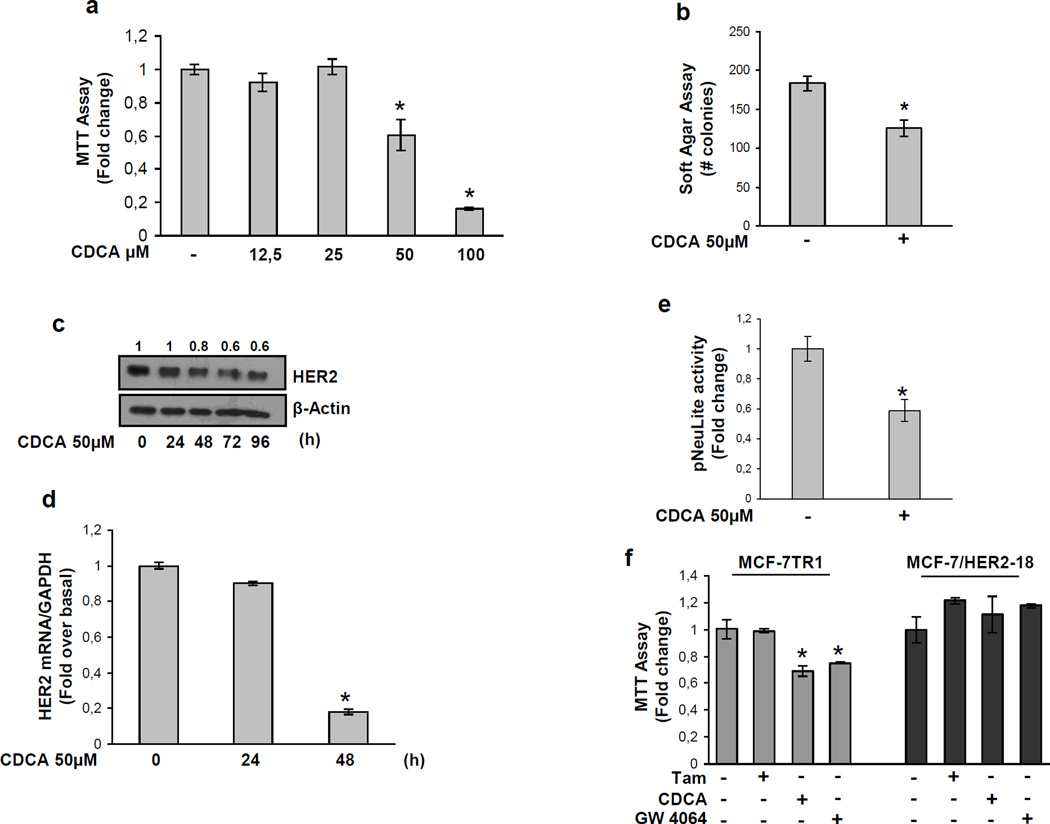
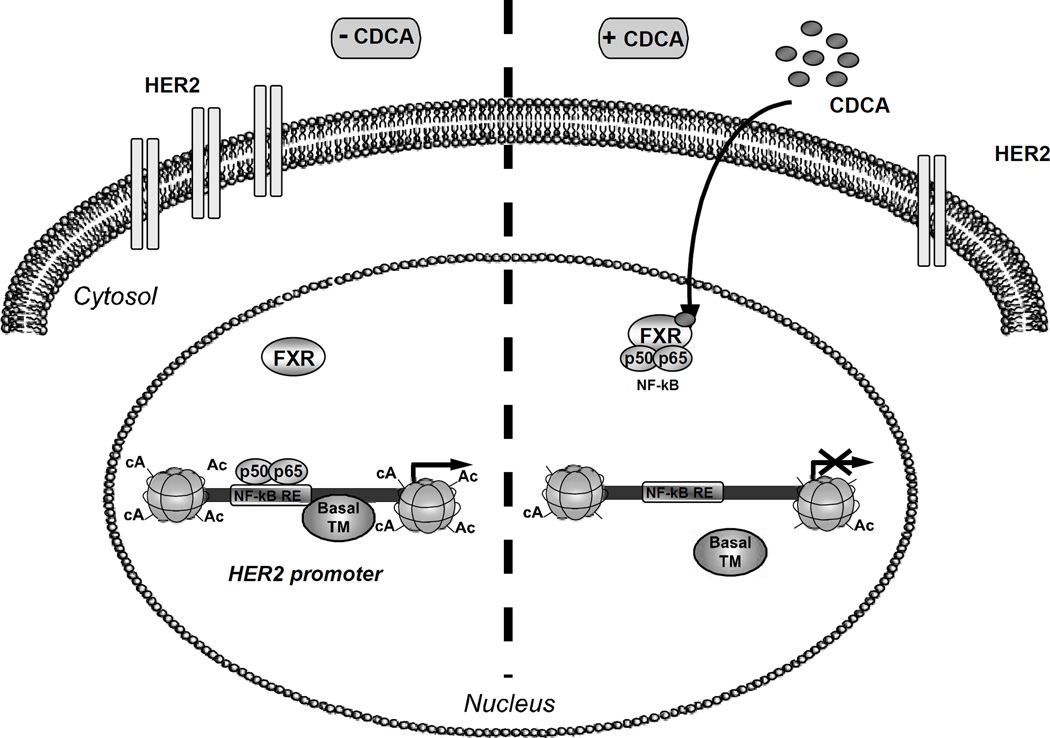
Tamoxifen (Tam) treatment is a first-line endocrine therapy for estrogen receptor-α-positive breast cancer patients. Unfortunately, resistance frequently occurs and is often related with overexpression of the membrane tyrosine kinase receptor HER2. This is the rationale behind combined treatments with endocrine therapy and novel inhibitors that reduce HER2 expression and signaling and thus inhibit Tam-resistant breast cancer cell growth. In this study, we show that activation of farnesoid X receptor (FXR), by the primary bile acid chenodeoxycholic acid (CDCA) or the synthetic agonist GW4064, inhibited growth of Tam-resistant breast cancer cells (termed MCF-7 TR1), which was used as an in vitro model of acquired Tam resistance. Our results demonstrate that CDCA treatment significantly reduced both anchorage-dependent and anchorage-independent epidermal growth factor (EGF)-induced growth in MCF-7 TR1 cells. Furthermore, results from western blot analysis and real-time reverse transcription-PCR revealed that CDCA treatment reduced HER2 expression and inhibited EGF-mediated HER2 and p42/44 mitogen-activated protein kinase (MAPK) phosphorylation in these Tam-resistant breast cancer cells. Transient transfection experiments, using a vector containing the human HER2 promoter region, showed that CDCA treatment downregulated basal HER2 promoter activity. This occurred through an inhibition of nuclear factor-κB transcription factor binding to its specific responsive element located in the HER2 promoter region as revealed by mutagenesis studies, electrophoretic mobility shift assay and chromatin immunoprecipitation analysis. Collectively, these data suggest that FXR ligand-dependent activity, blocking HER2/MAPK signaling, may overcome anti-estrogen resistance in human breast cancer cells and could represent a new therapeutic tool to treat breast cancer patients that develop resistance.
Figures
Similar articles
-
Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer.J Natl Cancer Inst. 2004 Jun 16;96(12):926-35. doi: 10.1093/jnci/djh166. J Natl Cancer Inst. 2004. PMID: 15199112
-
[Involvement of epidermal growth factor receptor signaling pathway in tamoxifen resistance of MCF-7 cells].Ai Zheng. 2006 Jul;25(7):839-43. Ai Zheng. 2006. PMID: 16831274 Chinese.
-
Human epidermal growth factor receptor 2 status modulates subcellular localization of and interaction with estrogen receptor alpha in breast cancer cells.Clin Cancer Res. 2004 Jun 1;10(11):3621-8. doi: 10.1158/1078-0432.CCR-0740-3. Clin Cancer Res. 2004. PMID: 15173068
-
Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer.Endocr Relat Cancer. 2005 Jul;12 Suppl 1:S99-S111. doi: 10.1677/erc.1.01005. Endocr Relat Cancer. 2005. PMID: 16113104 Review.
-
Inhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer.Clin Cancer Res. 2001 Dec;7(12 Suppl):4436s-4442s; discussion 4411s-4412s. Clin Cancer Res. 2001. PMID: 11916237 Review.
Cited by
-
Farnesoid X receptor: a potential therapeutic target in multiple organs.Histol Histopathol. 2020 Dec;35(12):1403-1414. doi: 10.14670/HH-18-301. Epub 2021 Jan 4. Histol Histopathol. 2020. PMID: 33393073 Review.
-
The role of bile acids in reducing the metabolic complications of obesity after bariatric surgery: a systematic review.Int J Obes (Lond). 2015 Nov;39(11):1565-74. doi: 10.1038/ijo.2015.115. Epub 2015 Jun 17. Int J Obes (Lond). 2015. PMID: 26081915
-
Expression signatures and roles of microRNAs in inflammatory breast cancer.Cancer Cell Int. 2019 Jan 31;19:23. doi: 10.1186/s12935-018-0709-6. eCollection 2019. Cancer Cell Int. 2019. PMID: 30733644 Free PMC article. Review.
-
Src-mediated cross-talk between farnesoid X and epidermal growth factor receptors inhibits human intestinal cell proliferation and tumorigenesis.PLoS One. 2012;7(10):e48461. doi: 10.1371/journal.pone.0048461. Epub 2012 Oct 31. PLoS One. 2012. PMID: 23119029 Free PMC article.
-
Heat shock proteins and heat shock factor 1 in carcinogenesis and tumor development: an update.Arch Toxicol. 2013 Jan;87(1):19-48. doi: 10.1007/s00204-012-0918-z. Epub 2012 Aug 11. Arch Toxicol. 2013. PMID: 22885793 Free PMC article. Review.
References
-
- Allred DC, Clark GM, Molina R, Tandon AK, Schnitt SJ, Gilchrist KW, et al. Overexpression of HER-2/neu and its relationship with other prognostic factors change during the progression of in situ to invasive breast cancer. Hum Pathol. 1992;23:974–979. - PubMed
-
- Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276:28857–28865. - PubMed
-
- Arpino G, Green SJ, Allred DC, Lew D, Martino S, Osborne CK, et al. HER-2 amplification, HER-1 expression, and tamoxifen response in estrogen receptor-positive metastatic breast cancer: a southwest oncology group study. Clin Cancer Res. 2004;10:5670–5676. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
