

Lysosomal lipid storage diseases
- PMID: 21502308
- PMCID: PMC3098676
- DOI: 10.1101/cshperspect.a004804
Lysosomal lipid storage diseases
Abstract
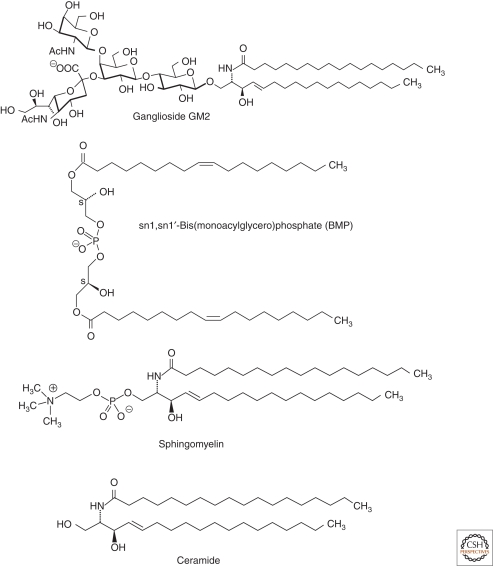
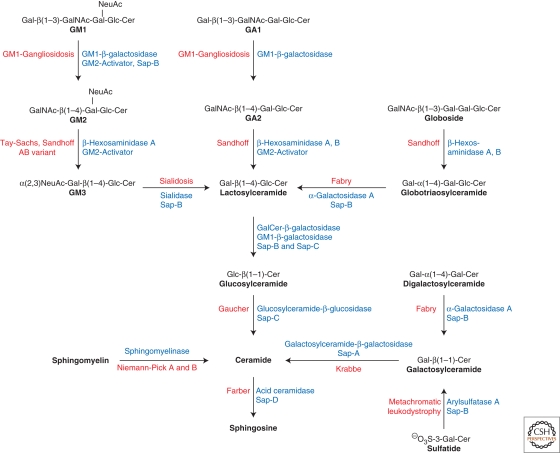
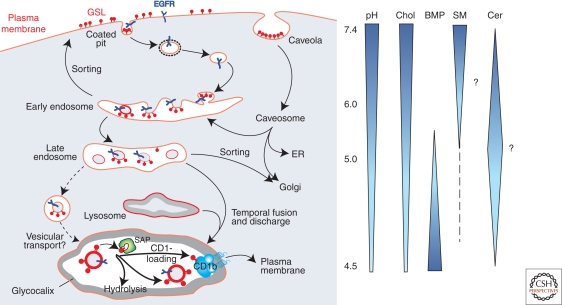
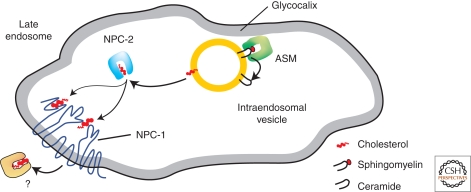
Lysosomal lipid storage diseases, or lipidoses, are inherited metabolic disorders in which typically lipids accumulate in cells and tissues. Complex lipids, such as glycosphingolipids, are constitutively degraded within the endolysosomal system by soluble hydrolytic enzymes with the help of lipid binding proteins in a sequential manner. Because of a functionally impaired hydrolase or auxiliary protein, their lipid substrates cannot be degraded, accumulate in the lysosome, and slowly spread to other intracellular membranes. In Niemann-Pick type C disease, cholesterol transport is impaired and unesterified cholesterol accumulates in the late endosome. In most lysosomal lipid storage diseases, the accumulation of one or few lipids leads to the coprecipitation of other hydrophobic substances in the endolysosomal system, such as lipids and proteins, causing a "traffic jam." This can impair lysosomal function, such as delivery of nutrients through the endolysosomal system, leading to a state of cellular starvation. Therapeutic approaches are currently restricted to mild forms of diseases with significant residual catabolic activities and without brain involvement.
Figures
Similar articles
-
A Comparative Study on the Alterations of Endocytic Pathways in Multiple Lysosomal Storage Disorders.Mol Pharm. 2016 Feb 1;13(2):357-368. doi: 10.1021/acs.molpharmaceut.5b00542. Epub 2016 Jan 11. Mol Pharm. 2016. PMID: 26702793 Free PMC article.
-
Sphingolipids and lysosomal pathologies.Biochim Biophys Acta. 2014 May;1841(5):799-810. doi: 10.1016/j.bbalip.2013.10.015. Epub 2013 Oct 31. Biochim Biophys Acta. 2014. PMID: 24184515 Review.
-
Finding pathogenic commonalities between Niemann-Pick type C and other lysosomal storage disorders: Opportunities for shared therapeutic interventions.Biochim Biophys Acta Mol Basis Dis. 2020 Oct 1;1866(10):165875. doi: 10.1016/j.bbadis.2020.165875. Epub 2020 Jun 6. Biochim Biophys Acta Mol Basis Dis. 2020. PMID: 32522631 Review.
-
Complex lipid trafficking in Niemann-Pick disease type C.J Inherit Metab Dis. 2015 Jan;38(1):187-99. doi: 10.1007/s10545-014-9794-4. Epub 2014 Nov 26. J Inherit Metab Dis. 2015. PMID: 25425283 Review.
-
The expanding boundaries of sphingolipid lysosomal storage diseases; insights from Niemann-Pick disease type C.Biochem Soc Trans. 2023 Oct 31;51(5):1777-1787. doi: 10.1042/BST20220711. Biochem Soc Trans. 2023. PMID: 37844193 Free PMC article. Review.
Cited by
-
Lysosomal physiology.Annu Rev Physiol. 2015;77:57-80. doi: 10.1146/annurev-physiol-021014-071649. Annu Rev Physiol. 2015. PMID: 25668017 Free PMC article. Review.
-
Sphingolipid lysosomal storage diseases: from bench to bedside.Lipids Health Dis. 2021 May 3;20(1):44. doi: 10.1186/s12944-021-01466-0. Lipids Health Dis. 2021. PMID: 33941173 Free PMC article. Review.
-
Membrane lipids regulate ganglioside GM2 catabolism and GM2 activator protein activity.J Lipid Res. 2015 Sep;56(9):1747-61. doi: 10.1194/jlr.M061036. Epub 2015 Jul 14. J Lipid Res. 2015. PMID: 26175473 Free PMC article.
-
Sphingolipid metabolic pathway: an overview of major roles played in human diseases.J Lipids. 2013;2013:178910. doi: 10.1155/2013/178910. Epub 2013 Aug 4. J Lipids. 2013. PMID: 23984075 Free PMC article.
-
Genetic defects in the sphingolipid degradation pathway and their effects on microglia in neurodegenerative disease.Cell Signal. 2021 Feb;78:109879. doi: 10.1016/j.cellsig.2020.109879. Epub 2020 Dec 6. Cell Signal. 2021. PMID: 33296739 Free PMC article. Review.
References
-
- Amidon B, Brown A, Waite M 1996. Transacylase phospholipases in the synthesis of bis(monoacylglycero)phosphate. Biochemistry 35: 13995–14002 - PubMed
-
- Assmann G, Seedorf U 2001. Acid lipase deficiency: Wolman disease and cholesteryl ester storage disease. In The metabolic and molecular bases of inherited disease (ed. Scriver C.R., et al.), pp. 3551–3572 McGraw-Hill, New York
-
- Aslanidis C, Ries S, Fehringer P, Büchler C, Klima H, Schmitz G 1996. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics 33: 85–93 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources