

A case of pseudohypoaldosteronism type 1 with a mutation in the mineralocorticoid receptor gene
- PMID: 21503203
- PMCID: PMC3077507
- DOI: 10.3345/kjp.2011.54.2.90
A case of pseudohypoaldosteronism type 1 with a mutation in the mineralocorticoid receptor gene
Abstract
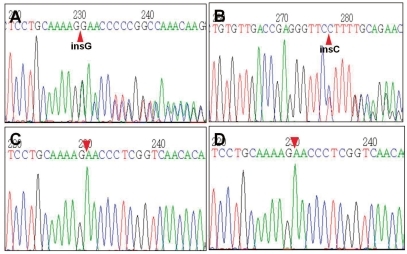
Pseudohypoaldosteronism type 1 (PHA1) is a rare form of mineralocorticoid resistance characterized in newborns by salt wasting with dehydration, hyperkalemia and failure to thrive. This disease is heterogeneous in etiology and includes autosomal dominant PHA1 owing to mutations of the NR3C2 gene encoding the mineralocorticoid receptor, autosomal recessive PHA1 due to mutations of the epithelial sodium channel (ENaC) gene, and secondary PHA1 associated with urinary tract diseases. Amongst these diseases, autosomal dominant PHA1 shows has manifestations restricted to renal tubules including a mild salt loss during infancy and that shows a gradual improvement with advancing age. Here, we report a neonatal case of PHA1 with a NR3C2 gene mutation (a heterozygous c.2146_2147insG in exon 5), in which the patient showed failure to thrive, hyponatremia, hyperkalemia, and elevated plasma renin and aldosterone levels. This is the first case of pseudohypoaldosteronism type 1 confirmed by genetic analysis in Korea.
Keywords: Infant; Mineralocorticoid; NR3C2 gene; Pseudohypoaldosteronism; Receptor.
Figures
References
-
- Sartorato P, Khaldi Y, Lapeyraque AL, Armanini D, Kuhnle U, Salomon R, et al. Inactivating mutations of the mineralocorticoid receptor in Type I pseudohypoaldosteronism. Mol Cell Endocrinol. 2004;217:119–125. - PubMed
-
- Geller DS. Mineralocorticoid resistance. Clin Endocrinol (Oxf) 2005;62:513–520. - PubMed
-
- Hanukoglu A. Type I pseudohypoaldosteronism includes two clinically and genetically distinct entities with either renal or multiple target organ defects. J Clin Endocrinol Metab. 1991;73:936–944. - PubMed
-
- Geller DS, Rodriguez-Soriano J, Vallo Boado A, Schifter S, Bayer M, Chang SS, et al. Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type I. Nat Genet. 1998;19:279–281. - PubMed
-
- Kwon YS, Shin HG, Ahn MS, Kim HB. A case of pseudohypoaldosteronism. J Korean Pediatr Soc. 1992;35:984–988.
LinkOut - more resources
Full Text Sources
