

Tumorigenic polyploid cells contain elevated ROS and ARE selectively targeted by antioxidant treatment
- PMID: 21503880
- PMCID: PMC3156849
- DOI: 10.1002/jcp.22793
Tumorigenic polyploid cells contain elevated ROS and ARE selectively targeted by antioxidant treatment
Abstract
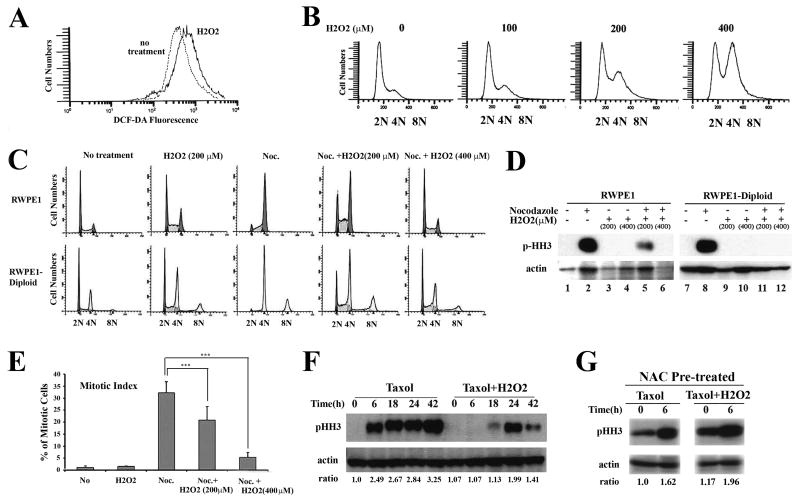
Polyploidy has been linked to tumorigenicity mainly due to the chromosomal aberrations. Elevated reactive oxygen species (ROS) generation, on the other hand, has also been associated with oncogenic transformation in most cancer cells. However, a possible link between ploidy and ROS is largely unexplored. Here we have examined the role of ROS in the tumorigenicity of polyploid cells. We show that polyploid prostate and mammary epithelial cells contain higher levels of ROS due to their higher mitochondrial contents. ROS levels and mitochondrial mass are also higher in dihydrocytochalasin B (DCB)-induced polyploid cells, suggesting that higher levels of ROS observed in polyploid cell can occur due to cytokinesis failure. Interestingly, polyploid cells were more sensitive to the inhibitory effect of the antioxidant, N-Acetyl-L-cysteine (NAC), than control diploid cells. Treatment of polyploid/diploid cells with NAC led to the selective elimination of polyploid cells over time and abrogated the tumorigenicity of polyploid cells. This effect was partially mediated via the Akt signaling pathway. We next explored a possible role for ROS in promoting chromosomal instability by analyzing the effects of ROS on the mitotic stage of the cell cycle. Enhancing ROS levels by treating cells with hydrogen peroxide delayed not only entry into and but also exit from mitosis. Furthermore, increasing ROS levels significantly increased taxol resistance. Our results indicated that increased ROS in polyploid cells can contribute to tumorigenicity and highlight the therapeutic potential of antioxidants by selectively targeting the tumorigenic polyploid cells and by reversing taxol resistance.
Copyright © 2011 Wiley Periodicals, Inc.
Figures






Similar articles
-
A role for polyploidy in the tumorigenicity of Pim-1-expressing human prostate and mammary epithelial cells.PLoS One. 2008 Jul 2;3(7):e2572. doi: 10.1371/journal.pone.0002572. PLoS One. 2008. PMID: 18596907 Free PMC article.
-
N-acetylcysteine modulates the cytotoxic effects of Paclitaxel.Chemotherapy. 2011;57(4):298-304. doi: 10.1159/000329510. Epub 2011 Jul 20. Chemotherapy. 2011. PMID: 21778718
-
The cytoprotective effect of N-acetyl-L-cysteine against ROS-induced cytotoxicity is independent of its ability to enhance glutathione synthesis.Toxicol Sci. 2011 Mar;120(1):87-97. doi: 10.1093/toxsci/kfq364. Epub 2010 Dec 6. Toxicol Sci. 2011. PMID: 21135414 Free PMC article.
-
TNF-alpha/cycloheximide-induced apoptosis in intestinal epithelial cells requires Rac1-regulated reactive oxygen species.Am J Physiol Gastrointest Liver Physiol. 2008 Apr;294(4):G928-37. doi: 10.1152/ajpgi.00219.2007. Epub 2008 Jan 24. Am J Physiol Gastrointest Liver Physiol. 2008. PMID: 18218673
-
The Antioxidant N-Acetylcysteine Inhibits Inflammatory and Apoptotic Processes in Human Conjunctival Epithelial Cells in a High-Glucose Environment.Invest Ophthalmol Vis Sci. 2015 Aug;56(9):5614-21. doi: 10.1167/iovs.15-16909. Invest Ophthalmol Vis Sci. 2015. PMID: 26305534
Cited by
-
Polyploid giant cancer cells: origin, possible pathways of formation, characteristics, and mechanisms of regulation.Front Cell Dev Biol. 2024 Jul 11;12:1410637. doi: 10.3389/fcell.2024.1410637. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 39055650 Free PMC article. Review.
-
RAMP1 is a direct NKX3.1 target gene up-regulated in prostate cancer that promotes tumorigenesis.Am J Pathol. 2013 Sep;183(3):951-63. doi: 10.1016/j.ajpath.2013.05.021. Epub 2013 Jul 16. Am J Pathol. 2013. PMID: 23867798 Free PMC article.
-
An Overview of Oxidative Stress in Sex Chromosome Aneuploidies in Pediatric Populations.Antioxidants (Basel). 2025 Apr 29;14(5):531. doi: 10.3390/antiox14050531. Antioxidants (Basel). 2025. PMID: 40427413 Free PMC article. Review.
-
Metabolism-induced oxidative stress and DNA damage selectively trigger genome instability in polyploid fungal cells.EMBO J. 2019 Oct 1;38(19):e101597. doi: 10.15252/embj.2019101597. Epub 2019 Aug 26. EMBO J. 2019. PMID: 31448850 Free PMC article.
-
Single-cell morphological and transcriptome analysis unveil inhibitors of polyploid giant breast cancer cells in vitro.Commun Biol. 2023 Dec 21;6(1):1301. doi: 10.1038/s42003-023-05674-5. Commun Biol. 2023. PMID: 38129519 Free PMC article.
References
-
- Barnouin K, Dubuisson ML, Child ES, Fernandez de Mattos S, Glassford J, Medema RH, Mann DJ, Lam EW. H2O2 induces a transient multi-phase cell cycle arrest in mouse fibroblasts through modulating cyclin D and p21Cip1 expression. J Biol Chem. 2002;277(16):13761–13770. - PubMed
-
- Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci. 2008;121(1):1–84. - PubMed
-
- Castillo A, Morse HC, 3rd, Godfrey VL, Naeem R, Justice MJ. Overexpression of Eg5 causes genomic instability and tumor formation in mice. Cancer Res. 2007;67(21):10138–10147. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
