

Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems
- PMID: 21504589
- PMCID: PMC3112146
- DOI: 10.1186/1471-2164-12-199
Using RNA-Seq for gene identification, polymorphism detection and transcript profiling in two alfalfa genotypes with divergent cell wall composition in stems
Abstract
Background: Alfalfa, [Medicago sativa (L.) sativa], a widely-grown perennial forage has potential for development as a cellulosic ethanol feedstock. However, the genomics of alfalfa, a non-model species, is still in its infancy. The recent advent of RNA-Seq, a massively parallel sequencing method for transcriptome analysis, provides an opportunity to expand the identification of alfalfa genes and polymorphisms, and conduct in-depth transcript profiling.
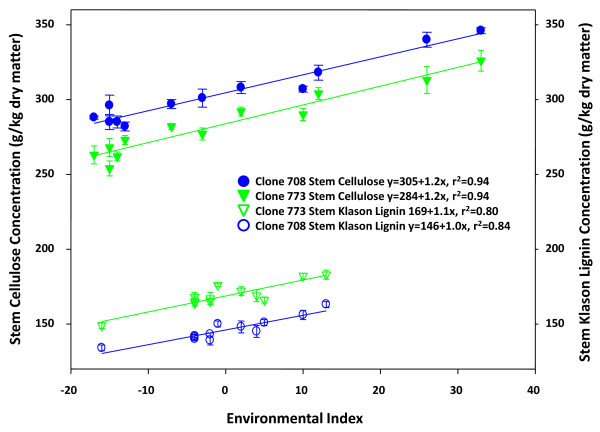
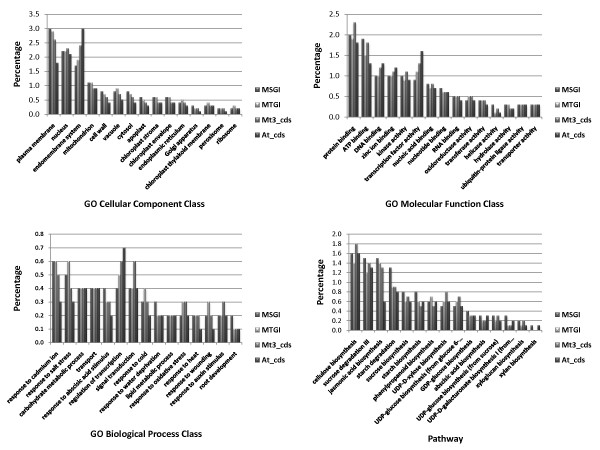
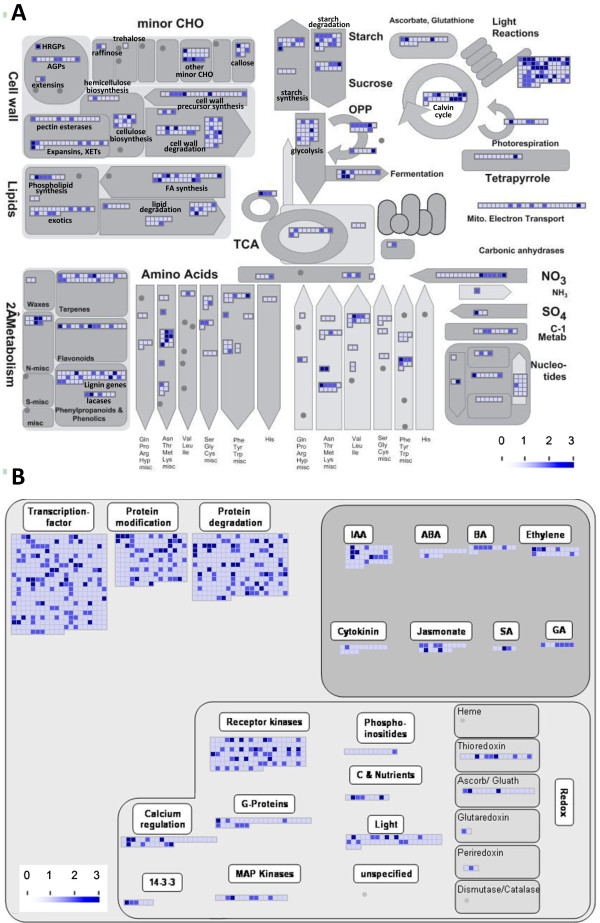
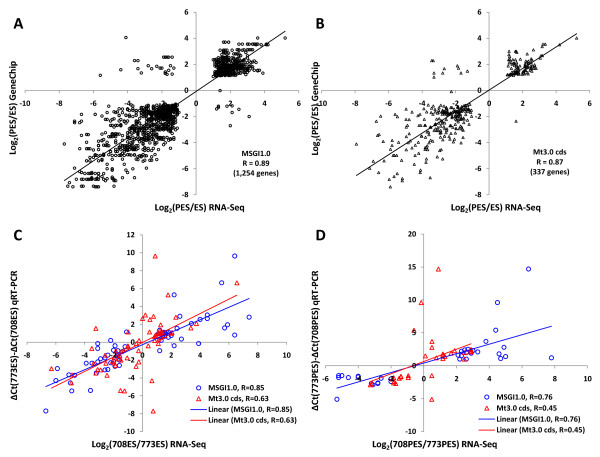
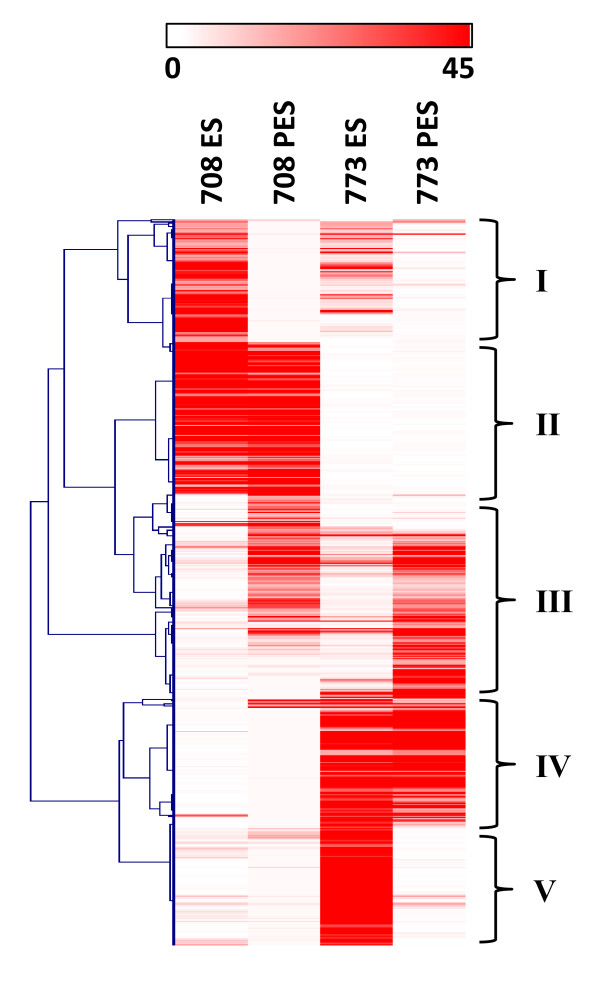
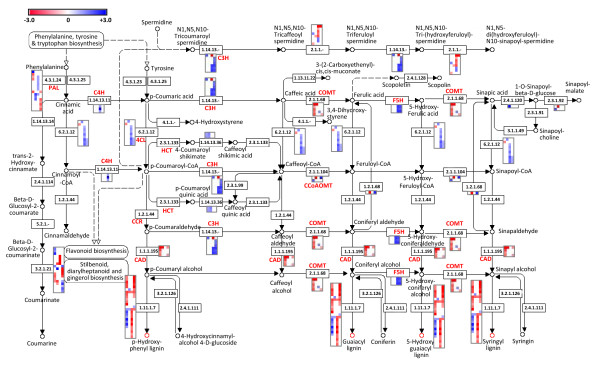
Results: Cell walls in stems of alfalfa genotype 708 have higher cellulose and lower lignin concentrations compared to cell walls in stems of genotype 773. Using the Illumina GA-II platform, a total of 198,861,304 expression sequence tags (ESTs, 76 bp in length) were generated from cDNA libraries derived from elongating stem (ES) and post-elongation stem (PES) internodes of 708 and 773. In addition, 341,984 ESTs were generated from ES and PES internodes of genotype 773 using the GS FLX Titanium platform. The first alfalfa (Medicago sativa) gene index (MSGI 1.0) was assembled using the Sanger ESTs available from GenBank, the GS FLX Titanium EST sequences, and the de novo assembled Illumina sequences. MSGI 1.0 contains 124,025 unique sequences including 22,729 tentative consensus sequences (TCs), 22,315 singletons and 78,981 pseudo-singletons. We identified a total of 1,294 simple sequence repeats (SSR) among the sequences in MSGI 1.0. In addition, a total of 10,826 single nucleotide polymorphisms (SNPs) were predicted between the two genotypes. Out of 55 SNPs randomly selected for experimental validation, 47 (85%) were polymorphic between the two genotypes. We also identified numerous allelic variations within each genotype. Digital gene expression analysis identified numerous candidate genes that may play a role in stem development as well as candidate genes that may contribute to the differences in cell wall composition in stems of the two genotypes.
Conclusions: Our results demonstrate that RNA-Seq can be successfully used for gene identification, polymorphism detection and transcript profiling in alfalfa, a non-model, allogamous, autotetraploid species. The alfalfa gene index assembled in this study, and the SNPs, SSRs and candidate genes identified can be used to improve alfalfa as a forage crop and cellulosic feedstock.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
