

Lysosomal storage disorders: molecular basis and laboratory testing
- PMID: 21504867
- PMCID: PMC3500170
- DOI: 10.1186/1479-7364-5-3-156
Lysosomal storage disorders: molecular basis and laboratory testing
Abstract
Lysosomal storage disorders (LSDs) are a large group of more than 50 different inherited metabolic diseases which, in the great majority of cases, result from the defective function of specific lysosomal enzymes and, in few cases, of non-enzymatic lysosomal proteins or non-lysosomal proteins involved in lysosomal biogenesis. The progressive lysosomal accumulation of undegraded metabolites results in generalised cell and tissue dysfunction, and, therefore, multi-systemic pathology. Storage may begin during early embryonic development, and the clinical presentation for LSDs can vary from an early and severe phenotype to late-onset mild disease. The diagnosis of most LSDs--after accurate clinical/paraclinical evaluation, including the analysis of some urinary metabolites--is based mainly on the detection of a specific enzymatic deficiency. In these cases, molecular genetic testing (MGT) can refine the enzymatic diagnosis. Once the genotype of an individual LSD patient has been ascertained, genetic counselling should include prediction of the possible phenotype and the identification of carriers in the family at risk. MGT is essential for the identification of genetic disorders resulting from non-enzymatic lysosomal protein defects and is complementary to biochemical genetic testing (BGT) in complex situations, such as in cases of enzymatic pseudodeficiencies. Prenatal diagnosis is performed on the most appropriate samples, which include fresh or cultured chorionic villus sampling or cultured amniotic fluid. The choice of the test--enzymatic and/or molecular--is based on the characteristics of the defect to be investigated. For prenatal MGT, the genotype of the family index case must be known. The availability of both tests, enzymatic and molecular, enormously increases the reliability of the entire prenatal diagnostic procedure. To conclude, BGT and MGT are mostly complementary for post- and prenatal diagnosis of LSDs. Whenever genotype/phenotype correlations are available, they can be helpful in prognosis and in making decisions about therapy.
Figures
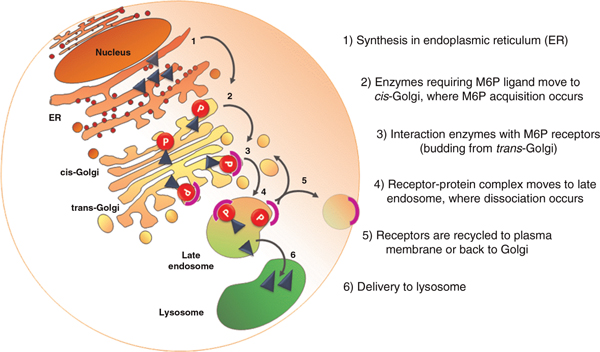
References
-
- Tay W. Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans Opthalmol Soc. 1881;1:55–57.
-
- Sachs B. On arrested cerebral development with special reference to cortical pathology. J Nerv Ment Dis. 1887;14:541–554.
-
- Gaucher PCE. De l'epithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leucemie. Academic thesis, Paris, France. 1882.
-
- Capper A, Epstein H, Schless RA. Gaucher's disease. Report of a case with presentation of a table differentiating the lipoid disturbances. Am J Med Sci. 1934;188:84. doi: 10.1097/00000441-193407000-00012. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
